
ABSTRACT
Selective catalytic reduction (SCR) catalysts are deactivated by several mineral and metallic trace elements at highly variable rates determined by fuel quality and furnace firing conditions. With a loss in activity, NO is reduced over a longer inlet length of the SCR monolith, which leaves a shorter trailing section to sustain the most favorable conditions to oxidize Hg0 and SO2. Since virtually no operating SCR was designed for Hg oxidation and since different monoliths are routinely combined as layers in particular units, the Hg oxidation performance of any SCR fleet is largely unmanaged. The analysis in this paper directly relates a measurement or manufacturer’s forecast on the deterioration in NO reduction with age to corresponding estimates for oxidation of Hg0. It accommodates any number of catalyst layers with grossly different properties, including materials from different manufacturers and different ages. In this paper, the analysis is applied to 16 full-scale SCRs in the Southern Company fleet to demonstrate that catalyst deactivation disrupts even the most prominent connections among the Hg0 oxidation performance of commercial SCRs and the behavior of fresh catalysts at lab, pilot, and even full scale.
Implications: Catalyst deactivation confounds even the most prominent connections among the Hg0 oxidation performance of commercial SCRs and the behavior of fresh catalyst at lab, pilot, and even full scale. The halogen dependence has been emphasized throughout the literature on catalytic Hg0 oxidation, based on a large database on fresh catalysts. But for deactivated catalysts in commercial SCRs, the number of layers is much more indicative of the Hg0 oxidation performance, in that SCRs with four layers perform better than those with three layers, and so on. The new qualified conclusion is that Hg0 oxidation is greater for progressively greater HCl concentrations only among SCRs with the same number of layers, even for an assortment of catalyst design specifications and operating conditions.
Introduction
Selective catalytic reduction (SCR) technology has become the technology of choice to meet stringent NOX emissions limits for coal-fired power plants. Along with NO reduction, SCR units also sustain Hg oxidation. The oxidation of elemental Hg (Hg0) into oxidized Hg (Hg2+) is important because only Hg2+ is water soluble and therefore amenable to capture in flue-gas desulfurization (FGD) scrubbing solutions. Extents of Hg0 oxidation of 90% or more across SCRs are routinely recorded in full-scale field tests (Chu et al., Citation2003), provided that the flue gas contains abundant Cl or Br species and that the SCR provides sufficient reactivity, favorable mass transport rates, and ample residence time for the chemistry to proceed to near-completion. When these conditions are satisfied, SCR/electrostatic precipitator (ESP)/FGD combinations are often the technology of choice for Hg control because they require no special operating procedures to lower Hg emissions, and may not require any specialized reagents. SCR catalysts also oxidize SO2 into SO3, and elevated SO3 levels can significantly exacerbate plume opacity problems once the SO3 condenses into fumes in the air preheater and beyond; SO3 also inhibits the capture of particulate-Hg on activated carbon sorbents and unburned carbon in fly ash, and thereby lowers ESP capture rates.
This paper demonstrates a simulation strategy to actively manage the multipollutant performance of an entire fleet of full-scale SCRs and its deterioration in time due to catalyst deactivation. The underlying performance model was introduced previously (Niksa and Freeman-Sibley, Citation2010a), and validated with extensive data sets on catalysts from four of the most popular SCR vendors in the American industry (Niksa and Freeman-Sibley, Citation2009, Citation2010b). It simultaneously describes the conversion of NO, Hg0, and SO2 along SCR catalyst channels in terms of the rates of film transport from the free stream to the external surface of a catalyst channel; diffusion of reactants through the internal catalyst pores; and the chemical conversion of the adsorbed participating species. In a succeeding study (Niksa et al., Citation2012), the catalyst channel model was expanded into a simulator for full-scale SCRs in which the properties of individual catalyst layers in a SCR monolith are completely independent, so that layers can have different geometrical specifications and ages, and catalysts from different vendors. The calculation sequence incorporates manufacturers’ activity ratios for NO reduction to describe how the NO reduction performance deteriorates with age.
This study uses SCR simulations for 16 full-scale SCRs in the Southern Company (SC) fleet to illustrate the impact of catalyst deactivation on the Hg0 oxidation performance, based on two limiting scenarios that relate the activities for NO reduction and Hg0 oxidation: one where they are proportionally diminished by deactivation and another in which only the NO reduction activity is diminished, which indirectly affects Hg0 oxidation. Pending a satisfactory validation of these predictions, the analysis enables regional plant managers to direct their resources toward the units that can deliver the best benefits, and to identify the most favorable strategies for the allocation of new catalysts within a particular SCR.
SCR unit simulations
Simulations for full-scale, multilayer SCR monoliths were developed from a custom computer package with three primary elements: (1) the SCR Catalyst Model, which describes the multipollutant performance along individual catalyst channels; (2) a calculation sequence that evaluates the SCR Catalyst Model in series for the properties of each successive catalyst layer in a SCR monolith; and (3) a catalyst deactivation algorithm, which reduces the reactivities for NO reduction and, perhaps, Hg0 oxidation according to a manufacturer’s activity at a specified age and the actual age of each catalyst layer. This section describes each element in turn.
Niksa and Fujiwara (Citation2005) introduced the first reaction mechanism for simultaneous NO reduction and Hg0 oxidation along SCRs in terms of lumped catalyst reactivities that resolved film transport of reactants onto the external catalyst channel wall from an overall chemical reactivity. The key features were a competition between NH3 and HCl for surface sites, and an Hg° oxidation reaction on chlorinated sites. This competitive adsorption subdivides the catalyst into two stages. An entry stage sustains NO reduction with high concentrations of adsorbed NH3, but the population of chlorinated sites remains very small as long as the HCl concentration is much lower than the NH3 concentration. This small population oxidizes relatively small proportions of Hg0. Once the NH3 has been consumed, a trailing stage is chlorinated much more extensively and therefore is able to rapidly oxidize Hg0. This mechanism interpreted extents of Hg0 oxidation by Cl species in lab-, pilot-, and full-scale SCR units for the complete domain of utility gas cleaning conditions. The strong coupling between NO reduction and Hg0 oxidation via competitive adsorption of NH3 and HCl was also independently validated with an extensive series of lab tests (Kamata et al., Citation2008). He et al. (Citation2009) observed HCl adsorption on monomeric vanadyl sites on a typical SCR catalyst, and suggested that such adsorbates explain the apparent inhibition of Hg0 oxidation in the presence of NH3.
Additional modeling work (Niksa and Freeman-Sibley, Citation2010a) expanded the original lumped analysis in three ways. First, simultaneous SO2 oxidation was incorporated; second, a conventional Thiele analysis now resolves internal pore diffusion across the catalyst wall thickness from the intrinsic reactivity of adsorbed species on the catalyst surface, to directly relate catalyst morphology and composition to the multipollutant performance of the SCR reactor; and, third, the analysis now accounts for simultaneous Hg0 oxidation by both Br and Cl species, to handle applications in which bromide salts are sprayed on coal or otherwise injected into a gas cleaning system. In the analysis, a simple premise connects NO and SO2 conversion to the Hg0 oxidation behavior on SCRs: that halogens compete for surface sites with NH3, and that Hg0 contacts these halogenated sites either from the gas phase or as a weakly adsorbed species, which is consistent with the virtual elimination of Hg0 adsorption on V2O5 by the DeNOX process (Staube et al., Citation2008). Since the extension to Br species was not included in the first publication on the distributed analysis for Hg0 oxidation, it is presented in the appendix. Although this feature was not invoked in any of the SCR simulations reported in this paper, extensive validations of this capability have been presented (Niksa et al., Citation2011).
The numerous reactivities and adsorption equilibrium relations were specified to quantitatively match the reported extents of Hg0 oxidation across broad domains of gas cleaning conditions (halogen levels, NH3/NO ratios, gas hourly space velocities [GHSV], temperatures) for catalysts from four major vendors at both the pilot and full scale (Niksa and Freeman-Sibley, Citation2009, Citation2010b). None of these parameters was adjusted in the present study.
Calculation sequence and catalyst deactivation protocol
In the simulations, the design specifications and age for each catalyst layer in the monolith are completely independent, so SCR monoliths may be constructed with layers from different manufacturers; with honeycomb or plate catalysts; with different geometrical specifications; and with different ages in each layer, as usually found among full-scale SCRs in service. The flue gas composition is specified at the SCR inlet, and then evaluated as a function of distance across each layer throughout the monolith.
The calculation sequence first uses the coal properties to assign a complete flue gas composition by varying the air flowrate to match a specified flue gas O2 concentration. The governing equations are then solved to determine extents of NO reduction, SO2 oxidation, and Hg0 oxidation along the multiple catalyst layers by accounting for their deactivation, according to the procedure outlined in the next subsection. The analysis restarts the boundary-layer growth at the inlet to each layer, which is important because mass transfer coefficients are nominally infinite at the inlet to every layer. Even though film transport rates rapidly decay as the boundary layer develops, the enhanced mass transfer at the layer inlets definitely boosts the performance of the whole SCR, and these enhancements are imposed at the beginning of every layer. The flue gas stream is assumed to be uniformly distributed across the faces into every layer, which ignores the flow maldistribution caused by inadequate flow conditioning upstream of the SCR, ash piling on the first layer, and ash plugging of some flow channels. The inlet NH3/NO ratio is also assumed to be uniform. Although such maldistribution effects can significantly degrade SCR performance if left unmanaged, they have little bearing on catalyst deactivation.
The conversion of Hg0 across an SCR is evaluated with an extent of Hg0 oxidation, SCRXHg0, is defined as follows:
where CHg0 represents the Hg0 concentrations at the inlet and outlet of the SCR monolith. The analogous conversion across a particular layer is denoted as IXHg0, where I is the layer index.
Operating conditions (inlet gas composition including NO and HCl levels; NH3/NO ratios; inlet Hg0 and Hg2+ levels; GHSV; temperatures) and design specifications (vendor; channel shape; pitch; wall thickness; porosities and mean pore sizes) were available for every layer of every SCR in the SC fleet. Deactivation histories estimated by the manufacturer were provided for every catalyst layer in the form of the initial NO reduction activity of the fresh catalyst (k0 in m/h) and the activity at the end of 16,000 hours of operation (kM). Catalyst operating hours at the time of the inventory were also reported. Catalyst manufacturers generally account for the ash loading of the primary coal feedstock in specifying the deactivation history, but whether or not they account for the expected poisons from each coal type is unknown. We used the reported operating hours for each layer and the manufacturer’s deactivation history to estimate the residual activity for age t in terms of the activity ratio, kd/k0, which is the ratio of the catalyst activity at any time, kd, to the initial catalyst activity, k0, given by the following formula:
where tM is 16,000 hr and t is the actual operating hours for the subject catalyst layer. For the oldest layers, this linear extrapolation gave very low or even negative residual activities, so the minimum activity ratio was limited to 0.4. This limit enabled plausible numerical simulations for all layer specifications in this study. Since even greater activity ratios gave very small conversions of NO and Hg0 across fully deactivated layers, this value does not restrict the simulation results in any way.
The activity ratio was then used to estimate the diminished extent of NOX reduction for the deactivated catalyst, according to
where α is the NH3/NO ratio at the inlet; IXNO,0 is the reference NO conversion across layer I for fresh catalyst; and IXNO,d is the estimate for NO reduction across the deactivated layer. Then the reactivity for NO reduction in the SCR Catalyst Model was adjusted to match the predicted NO reduction across the layer to IXNO,d.
Several gaseous species of As, P, K, Na, Ca, Pb, Cu, and others have been identified as poisons for SCR catalysts, although As, P, and Ca are the worst in most coal-burning utility applications. Solid compounds such as gypsum, ammonium bisulfate, and aerosol salts of Na, K, and other alkaline earth metals deactivate SCR catalysts by fouling and pore blocking. Aerosol salts of Ca and, to a lesser extent, Na and K generally block the catalyst pores and prevent the adsorption of NH3 and halogens, while oxides of As and P react with surface V−OH and V=O sites and deactivate them. Although deactivation mechanisms like pore blocking will affect both NH3 and halogen adsorption and, consequently, NO reduction and Hg0 oxidation, chemical deactivation will definitely affect NO reduction but not necessarily Hg0 oxidation.
Unfortunately, information in the literature is insufficient to assess the contribution for each deactivation mechanism to the deactivation of Hg0 oxidation. Eswaran and Stenger (Citation2008) reported a 30% reduction in the extents of Hg0 oxidation with 5 to 35 ppm HCl after 3300 hr in service with a commercial honeycomb catalyst. Kamata and Yukimura (Citation2012) evaluated a catalyst wall after 65,000 hr in service with bituminous coal that was deactivated by a continuous mask of Al/Si over the external surface, and found proportionate reductions in NO and Hg0 activities, as expected. But the findings for this atypical deactivation mode cannot be generalized to other coals. Li et al. (Citation2012) evaluated Hg0 oxidation efficiencies (but not DeNOX efficiencies) on an unspecified commercial SCR catalyst upon installation and after 40,000 hr of exposure to coal-derived flue gas. The reduction in the extent of Hg0 oxidation by one-third was qualitatively attributed to sintering and the greater loadings of Na, Ca, and Mg on the deactivated catalyst. Reported deactivation ratios (cf. eq 2) for Hg0 oxidation in commercial SCRs are roughly proportional to those for DeNOX (Defrancesco et al., Citation2012; Nakamoto et al., Citation2013; Reeves et al., Citation2013), but this relation does not distinguish the deactivation type.
In light of the current ambiguities surrounding how deactivation affects Hg0 oxidation, deactivation was examined with two limiting scenarios for pore blocking alone and for simultaneous pore blocking and chemical deactivation. In “Type I” deactivation, the deactivation mechanisms presumably prevent active sites from reacting with both NH3/NO and HCl/Hg0 species, and thereby diminish the site population. Smaller site populations necessarily support smaller surface coverages of the adsorbate species required for NO reduction and Hg0 oxidation. Since the reactivities for both NO reduction and Hg0 oxidation implicitly include the total population of active sites, both reactivities are diminished by a common multiplicative factor. The deactivation factor is specified to match the diminished extent of NO reduction for the deactivated catalyst from eq 3, shown earlier, and the same adjustment is applied to the Hg0 oxidation reactivity. The resulting reduction in Hg0 oxidation is a direct consequence of the lower Hg oxidation reactivity compounded by increased inhibition due to a greater concentration of NH3 further into the catalyst layer. Note that deactivation of NO reduction always sustains a greater NH3 concentration deeper into any catalyst layer, and greater NH3 concentrations inhibit Hg0 oxidation more strongly. Such compounding is important for Type I deactivation, but not for Type II deactivation.
The second deactivation scenario called “Type II” deactivation addresses situations where mechanisms prevent the active sites on the catalyst from reacting with NH3/NO but not with HCl/Hg0 species. Type II deactivation is implemented like the first type, except that only the NO reactivity is lowered to match IXNO,d for the estimated activity ratio.
Even though both deactivation scenarios are calibrated to the same IXNO,d, Type II deactivation exerts a weaker impact on Hg0 oxidation, because stronger NH3 inhibition is the only effect that comes into play. With Type I deactivation, Hg0 oxidation is diminished by the direct reduction in the Hg0 oxidation reactivity plus the stronger inhibition associated with the greater NH3 concentrations resulting from the lower reactivity for NO reduction. With Type II deactivation, there is no direct reduction in the Hg0 oxidation reactivity, so extents of Hg0 oxidation are diminished exclusively by the stronger NH3 inhibition for the lower NO reduction reactivity, which persists along a greater length of the monolith.
These two deactivation scenarios are intended to bound the as-yet poorly characterized relation between the relative deactivation rates for DeNOX and Hg0 oxidation. It is conceivable that Hg0 oxidation is deactivated faster than DeNOX and therefore beyond the scope of this analysis, but this is unlikely because none of the estimated deactivation rates for Hg0 oxidation have corroborated this rank ordering and, as seen in the following, the predicted impact of Type I deactivation on Hg0 oxidation is already very strong.
Subject SCR fleet
The simulations were based on detailed design specifications for 16 full-scale SCRs at nine power plants, of which seven fire bituminous coals and two fire PRB subbituminous coals. The inlet HCl concentrations varied from 4 to 99 ppmv, and inlet NO ranged from 92 to 457 ppm. None of the units were promoted with Br salts. The GHSVs varied between 1424 and 3076 hr−1. Most SCRs operated between 370 and 405°C, although this variation would only affect Hg0 oxidation in the hottest units (Niksa and Freeman-Sibley, Citation2009, Citation2010b). Most of the SCRs had four catalyst layers, some had three, and one had two layers. Most of the catalyst layers were honeycombs, although there were also plate monoliths from different vendors. The ages of the catalyst layers varied from 4,000 to 51,000 hr, which correspond to activity ratios from 0.94 to 0.4.
Stringent evaluations of the simulation results require data on the concentrations of NO or NH3 and Hg0 at the outlet of every layer of every SCR. However, none of these species were monitored within any of these SCRs, and only the NO concentrations were monitored at the SCR outlet. Unfortunately, such data cannot validate predicted values because outlet NO concentrations are always consistent with an extent of NO reduction based on the inlet NH3/NO ratio, to satisfy stipulations on NH3 slip.
Simulation results
Each simulation took less than 5 sec on a dual-core microprocessor operating at 1.86 GHz. The most important features of deactivation across multilayer monoliths are illustrated with simulations for an SCR with four honeycomb layers of identical design characteristics but ages that varied by a factor of two. The flue gas was generated with an Eastern bituminous coal. In simulations for the left panel of , all catalysts were fresh and therefore had an activity ratio of unity. The predicted axial profiles for NH3 and NO concentrations and NO and Hg0 conversions are plotted along a normalized length coordinate where 0.0 represents the entrance to the first layer and 1.0 represents the exit from the last layer. Since all four layers installed in this SCR are physically identical, a length increment of 0.25 represents the length of each catalyst layer. The interlayer distances are not shown.
Figure 1. Predicted profiles of (right y-axes) NH3 and NO concentrations, and (left y-axes) cumulative NO and Hg0 conversions across (left panel) fresh catalyst and (right panel) in-service catalysts whose ages varied by a factor of 2.
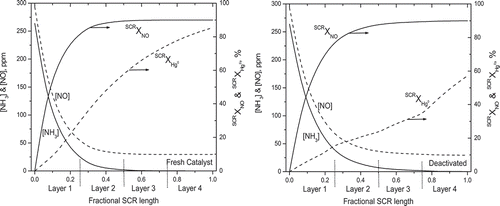
With fresh catalyst, the NO and NH3 concentrations drop sharply in the first layer with a corresponding increase in SCRXNO, and at the exit of the first layer, 81.5% of the inlet NO has been reduced. NO reduction continues in the next layer at a slower rate and quickly reaches the final asymptotic value, while NH3 is nearly eliminated near the outlet of the second layer. The remaining two layers provide minimal improvements to the NO reduction efficiency. The inhibitory effect of NH3 on Hg0 oxidation is clearly apparent in these results. At the midpoint of the first catalyst layer, where the NH3 concentration is still substantial, only 11% of the Hg0 was oxidized, whereas 62% of the NO had already been reduced. At the outlet of the first layer, Hg0 oxidation is just below 30% whereas NO reduction exceeds 81%. Once NH3 was depleted by the end of the first layer, Hg0 oxidation was determined by the HCl concentration, which remains constant because Hg0 conversion is too small in absolute terms to affect the halogen concentration. Consequently, extents of Hg0 oxidation across the three downstream layers are the same at approximately 40%, and the cumulative conversion of Hg0 steadily increases to about 85%. Although the fresh catalyst is overdesigned for NO reduction, a similar asymptotic behavior is not apparent for SCRXHg0 due to the high HCl concentration in this case.
The impact of Type I deactivation, which is the stronger of the two deactivation types, is evident in the right panel of . The most reactive layers are at the two ends, with activity ratios of 0.81, while catalysts with greater ages are in the middle with activity ratios of 0.69 and 0.63. Deactivation shifts the NH3 and NO profiles further into the monolith such that the first layer eliminates 77% of NO, versus 81.5% for fresh catalyst. This difference is modest because the first catalyst layer was relatively fresh. This relatively small difference in NO reduction is nevertheless sufficient to cut Hg0 oxidation in half, to about 16%, from 29% for fresh catalyst. Ammonia concentrations at the outlet of the first layer are 38 ppm for deactivated catalyst versus 25 ppm for fresh catalyst. More NH3 inhibits the adsorption of HCl and thereby indirectly diminishes Hg0 oxidation. Thus, the large difference in 1XHg0 is due to the compounding effect of the reduction in the reactivities for both NO and Hg0 conversion.
Since the SCR is overdesigned for NO reduction, SCRXNO reaches its asymptotic value toward the end of the third layer, despite strong deactivation in the second and third layers, and the simulations satisfy the limit on NH3 slip of 2 ppm. In contrast, Hg0 oxidation does not approach the uniform layer conversions for fresh catalyst until the relatively fresh final layer, and the ultimate SCRXHg0 is well below the performance of the fresh monolith. The Hg0 oxidation across the four layers was approximately 16, 9, 15, and 35%, respectively, amounting to SCRXHg0 of 57%, versus 85% for the fresh catalyst. Also, the ages of layers 1 and 4 are identical, yet the first layer oxidized only 16% of Hg0 whereas the fourth layer converted 35%, clearly demonstrating the strong inhibitory effect of NH3 in the first layer. Moreover, IXHg0 varied significantly across the different layers, which clearly demonstrates that layer-specific ages and activity ratios are required to obtain reasonably accurate predictions for overall SCR Hg0 oxidation efficiencies.
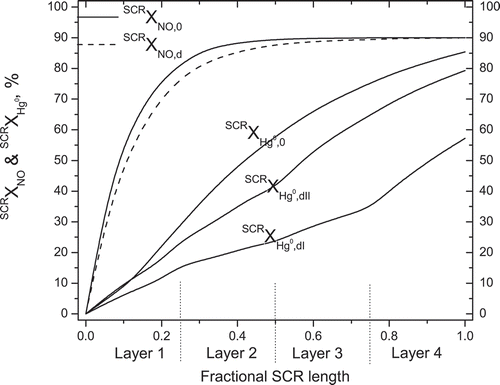
The cumulative NO reduction and Hg0 oxidation profiles for fresh catalyst (SCRXHg0,0), Type I deactivation (SCRXHg0,dI), and Type II deactivation (SCRXHg0,dII) are compared in . The NO reduction efficiencies at the SCR exit for the fresh and the deactivated catalysts are the same due to the overdesign of the SCR with respect to NO reduction, but the SCRXHg0 values are 85.4% for fresh catalyst, and 57 and 79% for deactivation of Types I and II, respectively. The substantial difference for these two deactivation scenarios underscores the importance of characterizing the different catalyst poisons and their associated deactivation mechanisms.
Figure 2. Predicted NO and Hg0 conversion profiles across the fresh (SCRXNO,0, SCRXHg0,0) and deactivated (SCRXNO,d, SCRXHg0,dI, SCRXHg0,dII) catalyst layers of the baseline SCR.
The next case compares four SCRs at the baseline power plant, each with four honeycomb layers of very similar design specifications but with ages that range from several thousand hours to several tens of thousands of hours. The sequences of activity ratios were 0.81, 0.69, 0.63, and 0.81 for Unit 1; 0.66, 0.66, 0.49, and 0.99 for Unit 2; 0.75, 0.40, 0.40, and 0.55 for Unit 3; and 0.40, 0.88, 0.40, and 0.63 for Unit 4. compares the cumulative extents of Hg0 oxidation for Type I deactivation of these four units. The SCRXHg0 values for the Type I deactivated catalysts, in descending order, are 57% at Unit 1; 51% at Unit 2; 44% at Unit 4; and 28% at Unit 3. This order corresponds to the sums of the operating hours of all layers in these units, which varied by a factor of 2. In , the order at the outlet is not the same as the orders at the outlets of each layer, because the ages of any particular layer are variable. For example, across the fourth layer at Unit 2, IXHg0 increased from 18 to 51% because this is the youngest layer of all, and also because NH3 was eliminated by the upstream layers. Conversely, the extents of Hg0 oxidation across layers 2 and 3 at Unit 3 and across layer 3 at Unit 4 are minimal because these layers are the oldest, and were assigned the minimum activity ratio of 0.4. Hence, the minimum value of the assigned activity ratio completely suppressed Hg0 oxidation, albeit under these particular operating conditions.
Figure 3. Predicted Hg0 oxidation profiles across the Type I deactivated SCR catalysts for the four units at the baseline power plant.
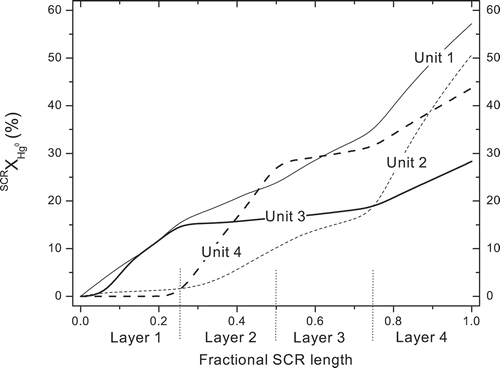
Strong NH3 inhibition is evident in the comparison of Hg0 oxidation across layers 1 and 4 of Units 1 and 4, because the two end layers at each of these units have the same ages and activity ratios but operate under the extremes in NH3 concentrations. At both units, IXHg0 values are greater by almost 20% across layer 4 than across layer 1. The implication is that a fresh catalyst layer should be added toward the end of the SCR to obtain the greatest benefit for Hg0 oxidation under the weakest inhibitory effect of NH3. However, the activities of the preceding layers play an important role, and better performance can sometimes be achieved by placing the freshest layer at intermediate positions.
The predictions for Type II deactivation follow these same trends except that the extents of Hg0 oxidation are consistently greater, as seen in .
The next case examines how deactivation affects the correlation between Hg0 oxidation and the flue gas HCl concentrations for the nine power plants in the SC fleet. Only one SCR was included from the plants with multiple SCRs. shows the predicted Hg0 oxidation for fresh catalysts and catalysts subject to Type I deactivation arranged in order of increasing Cl concentration from left to right. The HCl concentrations and the numbers of layers are also shown. The general trends in these predictions are the same for Type II deactivation.
Figure 4. The extents of Hg0 oxidation (hatched bars) for fresh catalyst (top panel) and deactivation (bottom panel) in order of increasing HCl concentrations (). The number of catalyst layers (▴) is plotted on the right y-axis.
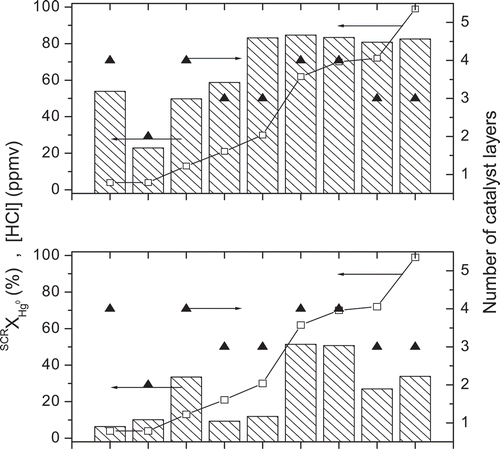
Except for one unit, the fresh catalyst layers exhibit a Cl dependence that is nominally first order through HCl concentrations to about 30 ppm, but then saturates to a negligible dependence for the greatest HCl concentrations. This trend is clearly apparent in the ranked Hg0 oxidation performance for fresh catalysts, and also in the large database in the literature for lab- and pilot-scale tests for fresh catalysts from many vendors. Accordingly, it is the expected form of the Cl dependence in this reaction system. However, the trend is thoroughly disrupted for the deactivated catalysts. Although more HCl is widely expected to give greater extents of Hg0 oxidation, the figure demonstrates that it is not the determining factor for deactivated catalysts. For deactivated catalysts, the number of layers is much more indicative of the Hg0 oxidation performance, in that SCRs with four layers perform better than those with three layers, with one exception whose HCl concentration is the lowest of all and whose NH3 level is relatively high, and whose layers are much older than the others. Similarly, the SCRs with three layers outperform the two-layer unit, with one exception. The expected Cl dependence is apparent among SCRs with the same number of layers; that is, Hg0 oxidation is greater for progressively greater Cl concentrations among SCRs with the same number of layers, even for an assortment of catalyst design specifications, operating conditions, and ages. However, the saturation to an asymptotic extent of Hg0 oxidation normally observed with 20 to 30 ppm HCl for fresh catalyst is shifted toward much greater HCl concentrations with deactivated catalysts.
Discussion
Catalyst deactivation confounds even the most prominent connections among the Hg0 oxidation performance of commercial SCRs and the behavior of fresh catalyst at lab, pilot, and even full scale. The halogen dependence has been emphasized throughout the literature on catalytic Hg0 oxidation, based on a large database on fresh catalysts. However, for deactivated catalysts in commercial SCRs, the number of layers is more indicative of the Hg0 oxidation performance, in that SCRs with four layers perform better than those with three layers, and so on. The qualified conclusion is that Hg0 oxidation is greater for progressively greater HCl concentrations among SCRs with the same number of layers, even for an assortment of catalyst design specifications and operating conditions. Even this conclusion is subject to the very large quantitative shift toward greater HCl concentrations in the apparent Cl dependence for deactivated catalysts, compared with the Cl dependence for fresh catalysts.
The SCRs in any large commercial fleet contain layers with markedly different catalyst properties; even when the properties are the same or similar, extents of deactivation are almost always different. Our simulations display, for the first time, remarkably large reductions in SCRXHg0 values due to catalyst deactivation, even while the SCRs continue to satisfy NH3 slip limits. The predicted impact of deactivation on Hg0 conversion was significant for every SCR in the SC fleet under both deactivation types. For Type I deactivation, the relative reductions in SCRXHg0 vary from 33 to 92 %, and the median reduction is 67%. For Type II deactivation, the relative reductions vary from 6 to 53%, and the median reduction is 25%. Suppose that at least 80% of Hg0 oxidation at every SCR in a fleet was needed to satisfy impending Hg regulatory requirements. According to , five units in the SC fleet would meet this demand with fresh catalyst, but in their actual catalyst condition under the worst deactivation scenario, only two would even remotely approach the threshold.
Of course, without stringent validations with field-test data, the simulation results are only suggestive. At a minimum, the relation between the impacts on NO reduction and Hg0 oxidation must be resolved to determine which of Types I and II deactivation is the more accurate scenario. But these simulations can only be validated with field-test data that monitor the NO reduction and Hg0 oxidation performance across all the layers in an SCR. Such data are now being recorded (Hinton et al., Citation2013 and Citation2014), although they are so expensive to obtain that it is unclear when or even whether the requisite database will ever be compiled. Whether Type I or Type II deactivation is the best scenario can be resolved with simultaneous monitoring of the efficiencies for NO reduction and Hg0 oxidation in any test at any scale. This requirement is only a modest supplement to testing protocols, although the issue needs to be addressed for all coal types that have distinctive poisoning potentials, and for all the major catalyst types.
For the time being, simulations based on distinctive characteristics for each layer provide the only means to factor the impact of catalyst deactivation into SCR management strategies for multipollutant control. One commercial catalyst vendor recently adopted this approach (Reeves et al., Citation2014), although the technical features of their simulations have not been disclosed.
Funding
This study was sponsored by Southern Company Services, Inc., and reflects its generous cooperation in providing detailed specifications and operating conditions for the SCRs in the simulations.
Additional information
Funding
Notes on contributors
Stephen Niksa
Stephen Niksa is president and founder of Niksa Energy Associates LLC in Belmont, CA.
Balaji Krishnakumar
Balaji Krishnakumar was a computational analyst at Niksa Energy Associates LLC.
Farrokh Ghoreishi
Farrokh Ghoreishi is the manager of SCR Operations at Southern Company Services.
References
- Chu, P., D. Laudal, L. Brickett, and C.W. Lee. 2003. Power plant evaluation of the effect of SCR technology on mercury. Proc. U.S. EPA-DoE-EPRI Combined Power Plant Air Pollutant Control Symp.: The MEGA Symp., Washington, DC, May 19–22.
- DiFrancesco, C.E., C.J. Bertole, J.T. Freeman, and K. Nochi. 2012. Achieving and maintaining Hg oxidation performance through SCR catalyst selection and management. Proc. U.S. EPA-DoE-EPRI Combined Power Plant Air Pollutant Control Symp.: The MEGA Symp., Baltimore, MD, August 28–31.
- Eswaran, S., and H.G. Stenger. 2008. Effect of halogens on mercury conversion in SCR catalysts. Fuel Process. Technol. 89:1153–59. doi:10.1016/j.fuproc.2008.05.007
- He, S., J. Zhou, Y. Zhu, Z. Luo, M. Ni, and K. Cen. 2009. Mercury oxidation over a vanadia-based selective catalytic reduction catalyst. Energy Fuels 23:253–59. doi:10.1021/ef800730f
- Hinton, S., T. Martz, and K. Cushing. 2013. Detailed SCR mercury speciation measurements supporting MATS mercury compliance. Proc. Air Quality IX, Arlington, VA, October 21–23.
- Hinton, S., T. Martz, C. Acharya, K. Cushing, and J. Paradis. 2014. SCR catalyst mercury oxidation behavior supporting MATS mercury compliance. Proc. U.S. EPA-DoE-EPRI Combined Power Plant Air Pollutant Control Symp.: The MEGA Symp., Baltimore, MD, August 19–21.
- Kamata, H., S.-I. Ueno, T. Naito, and A. Yukimura. 2008. Mercury oxidation over the V2O5(WO3)/TiO2 commercial SCR catalyst. Ind. Eng. Chem. Res. 47:8136. doi:10.1021/ie800363g
- Kamata, H., and A. Yukimura. 2012. Catalyst ageing in a coal combustion flue gas for mercury oxidation. Fuel Process. Technol. 104:295. doi:10.1016/j.fuproc.2012.05.025
- Li, J.-R., C. He, X.-S. Shang, J.-S. Chen, X.-W. Yu, and Y.-J. Yao. 2012. Oxidation efficiency of elemental mercury by SCR DeNOX catalysts. J. Fuel Chem. Technol. 40(2): 241–46. doi:10.1016/S1872-5813(12)60012-7
- Nakamoto, T., Y. Kato, Y. Nagai, and K. Neidig. 2013. SCR catalyst, a low-cost option for mercury mitigation. Proc. Air Quality IX, Arlington, VA, October 21–23.
- Niksa, S., and N. Fujiwara. 2005. A predictive mechanism for mercury oxidation on SCR catalysts under coal-derived flue gas. J. Air Waste Manage. Assoc. 56:1866–75. doi:10.1080/10473289.2005.10464779
- Niksa, S., and A. Freeman Sibley. 2009. Relating catalyst properties to the multipollutant performance of full-scale SCR systems. Int. Conf. on Air Quality VII, UND EERC, Arlington, VA, September 27–30.
- Niksa, S., and A. Freeman Sibley. 2010a. Relating catalyst properties to the multipollutant performance of full-scale SCR systems. Ind. Eng. Chem. Res. 49:6332–34.
- Niksa, S. and A. Freeman Sibley. 2010b. Relating catalyst properties to the multipollutant performance of full-scale SCR systems. EPRI-DOE-EPA-A&WMA Combined Utility Air Pollution Control Symposium: The MEGA Symp., Baltimore, MD, August 30–September 2.
- Niksa, S., B. Krishnakumar, and A. Freeman Sibley. 2011. Predicting the multipollutant performance of full-scale SCR systems with bromine addition. Int. Conf. on Air Quality VIII, UND EERC, Arlington, VA, October 26.
- Niksa, S., B. Krishnakumar, F. Ghoreishi, and C. Tyree. 2012. Analytical management of SCR catalyst lifetimes and multipollutant performance. EPRI-DOE-EPA-A&WMA Combined Utility Air Pollution Control Symposium: The MEGA Symp., Baltimore, MD, August 20–23.
- Reeves, C.T., C.J. Bertole, C.E. DiFrancesco, S.G. Pritchard, and K. Nochi. 2013. SCR catalyst selection and management for improved Hg oxidation performance. Proc. Air Quality IX, Arlington, VA, October 21–23, 2013.
- Reeves, C.T., C.J. Bertole, C.E. DiFrancesco, S.G. Pritchard, and K. Nochi. 2014. SCR catalyst selection and management for improved Hg oxidation performance. Proc. U. S. EPA-DoE-EPRI Combined Power Plant Air Pollutant Control Symp.: The MEGA Symp., Baltimore, MD, August 19–21.
- Staube, S., T. Hahn, and H. Koeser. 2008. Adsorption and oxidation of mercury in tail-end SCR DeNOX plants—Bench-scale investigations and speciation experiments. Appl. Catal. B Environ. 79:286–95.
Appendix: Expansion of the reaction mechanism for Hg0 oxidation by Br species
Based on compelling evidence from numerous studies on SCR catalysts, we previously adopted Eley–Rideal (ER) kinetics for both NO reduction and Hg0 oxidation, although there seems to be no detailed characterization work on kinetics for Hg0 oxidation by Br species on SCR catalysts. Based solely on the similar behavior of Cl and Br species under gas cleaning conditions, ER kinetics are implemented for Hg/Br chemistry as well. HBr and Br2 are assumed to form the same adsorbate on the catalyst, and all the halogenated adsorbates are completely independent.
The proposed rate law for Hg0 oxidation by adsorbed Br is therefore
where the fractional surface coverage of adsorbed Br is denoted by ΘBr. From the assumption that adsorbed NH3, Cl, and Br species are in adsorption equilibrium, it follows that the Hg0 oxidation rate is given by independent contributions for Cl and Br species, according to
where
Note that ΘCl and ΘBr grow as CSNH3 diminishes during the NO reduction, even though CHCl, CBr2, and CHBr in the free stream are uniform throughout the entire process. Also note that the term in square brackets that expresses the Br speciation will be a constant provided that the proportions of Br2 and HBr do not vary for different levels of Br addition. Pending empirical information on the relative adsorption rates of HBr and Br2, we fold this factor into an effective overall rate for Br species, and express the total amount of a Br addition as an equivalent level of HBr in flue gas.
When the new rate expression for Hg0 oxidation is inserted into the species conservation laws reported previously (Niksa and Freeman-Sibley 2010a), the following system of equations for Hg0 concentrations in the free-stream and at the external catalyst surface (ΦHg,G, ΦHg,S, and y = ΦHg0/ ΦHg,S) is obtained:
with an effectiveness factor given by
The analog to the Thiele modulus, Ω, is defined as
Otherwise, the length coordinate (Z⋆) and Sherwood number (Sh) have the definitions given previously (Niksa and Freeman-Sibley 2010a). In eq A.3c, XNH3 is the internal position in the catalyst wall where NH3 vanishes under some, but not all, conditions, which is given by a relation in the analysis for DeNOX.