
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Carlsbad Caverns National Park in southeastern New Mexico is adjacent to the Permian Basin, one of the most productive oil and gas regions in the country. The 2019 Carlsbad Caverns Air Quality Study (CarCavAQS) was designed to examine the influence of regional sources, including urban emissions, oil and gas development, wildfires, and soil dust on air quality in the park. Field measurements of aerosols, trace gases, and deposition were conducted from 25 July through 5 September 2019. Here, we focus on observations of fine particles and key trace gas precursors to understand the important contributing species and their sources and associated impacts on haze. Key gases measured included aerosol precursors, nitric acid and ammonia, and oil and gas tracer, methane. High-time resolution (6-min) PM2.5 mass ranged up to 31.8 µg m−3, with an average of 7.67 µg m−3. The main inorganic ion contributors were sulfate (avg 1.3 µg m−3), ammonium (0.30 µg m−3), calcium (Ca2+) (0.22 µg m−3), nitrate (0.16 µg m−3), and sodium (0.057 µg m−3). The WSOC concentration averaged 1.2 µg C m−3. Sharp spikes were observed in Ca2+, consistent with local dust generation and transport. Ion balance analysis and abundant nitric acid suggest PM2.5 nitrate often reflected reaction between nitric acid and sea salt, forming sodium nitrate, and between nitric acid and soil dust containing calcium carbonate, forming calcium nitrate. Sulfate and soil dust are the major contributors to modeled light extinction in the 24-hr average daily IMPROVE observations. Higher time resolution data revealed a maximum 1-hr extinction value of 90 Mm−1 (excluding coarse aerosol) and included periods of significant light extinction from BC as well as sulfate and soil dust. Residence time analysis indicated enrichment of sulfate, BC, and methane during periods of transport from the southeast, the direction of greatest abundance of oil and gas development.
Implications: Rapid development of U.S. oil and gas resources raises concerns about potential impacts on air quality in National Parks. Measurements in Carlsbad Caverns National Park provide new insight into impacts of unconventional oil and gas development and other sources on visual air quality in the park. Major contributors to visibility impairment include sulfate, soil dust (often reacted with nitric acid), and black carbon. The worst periods of visibility and highest concentrations of many aerosol components were observed during transport from the southeast, a region of dense Permian Basin oil and gas development.
Introduction
The development of unconventional oil and natural gas extraction techniques, including horizontal drilling and hydraulic fracturing, has unlocked significant new U.S. oil and natural gas (O/G) resources and led to a large increase in production from shale and tight oil and gas resources since 2000 (Brown, Lewis, and Weinberger Citation2015). Shale gas resources can be found in almost 30 U.S. states (U.S. EIA, Citation2021a). Tight oil production is driving U.S. oil production growth. The two main source regions are the Bakken shale play in the Northern Great Plains and the Wolfcamp shale play in the Permian Basin, located in west Texas and southeast New Mexico (United States Energy Information Administration (EIA) Citation2021a). Emissions of volatile organic compounds (VOCs) and nitrogen oxides (NOx) from oil and gas (O/G) operations have received attention due, in part, to their impacts on the production of ozone (Brown, Lewis, and Weinberger Citation2015; Field et al. Citation2015; Hecobian et al. Citation2019; Olaguer Citation2012; Prenni et al. Citation2016; Thompson, Hueber, and Helmig Citation2014). Less attention has been paid to the effects of O/G activities on particle concentrations and regional haze.
In this study, we examine the impact of O/G development and other sources on air quality in Carlsbad Caverns National Park (CAVE) in 2019. The park is located near extensive oil and natural gas development in the Permian Basin (). The major formation in this region is the Wolfcamp shale play (Gaswirth et al., Citation2018). This region is responsible for much of the shale gas production growth in the United States (U.S. EIA, Citation2021b). CAVE is in Eddy County, New Mexico, where 77% of SO2 and 56% of NOx emissions in the 2017 NEI are from petroleum and related industries. The next largest contributor is industrial fuel combustion for both SO2 and NOx, accounting for 17% and 23%, respectively. In neighboring counties (Lea NM, Culberson TX, Loving TX, and Reeves TX) to the south and east, an important upwind transport region during the study, petroleum and related industries accounted for 88% of SO2 emissions and 71% of NOx emissions (U.S. EPA, Citation2019). CAVE and the nearby town of Carlsbad frequently record high ozone values, often exceeding the 8-hr-average 70 ppbv National Ambient Air Quality Standard. A prior screening study of national parks in the southwestern U.S. (Benedict et al. Citation2020) revealed significant impacts of O/G emissions on VOC levels in CAVE, especially for light alkanes. The 2019 study in CAVE was motivated by these high ozone and VOC levels, along with concerns over sources of fine particle haze, and potential connection to a variety of regional sources including urban emissions, increased O/G development, wildfires, and erosion. Here, we focus primarily on aerosol concentrations in the park, their contributions to regional haze, and their sources. CAVE ozone and VOC levels will be discussed in a future publication.
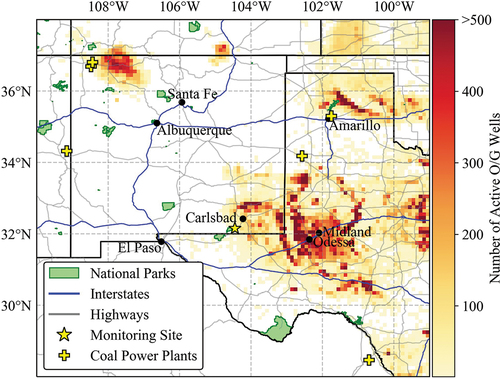
Figure 1. Active oil and natural gas wells near Carlsbad Caverns National Park (CAVE) (HIFLD, Citation2019). The color bar gives the number of active O/G wells in the given grid cell. The grid cell resolution is 0.1 degree by 0.1 degree. Cells with 500 or more active wells are shown with the same color. National Park boundaries are shown in green. Larger cities in the surrounding area are marked with black dots and labeled. The monitoring site, in CAVE is shown as a yellow star. Coal-fired power plants in the region are shown as yellow plus signs (U.S. EIA, Citation2022).
O/G operations and associated traffic and infrastructure can lead to direct emission of fine particulate matter (PM2.5) and of gaseous precursors that lead to secondary particle formation. Black carbon (BC) is emitted from incomplete combustion processes and is associated with O/G development and production, with emissions from flaring and diesel engine use (Bond et al. Citation2006; Khalek et al. Citation2015; Shen et al. Citation2021; Yanowitz, McCormick, and Graboski Citation2000). During flaring excess gas is burned and can yield significant emissions of NOx, BC, and methane (CH4) during incomplete combustion (Allen et al. Citation2016; Böttcher et al. Citation2021; Conrad and Johnson Citation2019; Weyant et al. Citation2016). BC is of particular interest because of its strong absorption of solar radiation, in addition to the general adverse health impacts of PM2.5 (Bond et al., Citation2013). Other organic aerosol species can also be emitted or formed by secondary processes/reactions producing secondary organic aerosol (Pandis and Seinfeld Citation2016).
Oil and gas development and production can also impact levels of sulfate, nitrate, and ammonium particulate species. Direct emission of sulfur dioxide (SO2) and NOx leads to the formation of sulfuric and nitric acids, respectively. Both can react with ammonia (NH3) to form hygroscopic particles with important implications for total PM and light scattering. Ammonium nitrate formation, in particular, has been tied to O/G operations in the western U.S. (Elvidge et al. Citation2009; Evanoski-Cole et al. Citation2017; Li et al. Citation2012; Prenni et al. Citation2016). The formation of ammonium sulfate ((NH4)2SO4) and ammonium nitrate (NH4NO3) is dependent on ammonia availability and thermodynamic conditions. If ammonia is very limited, sulfuric acid will exist in the particle phase. As ammonia availability increases, NH4HSO4 (ammonium bisulfate), (NH4)3H(SO4)2 (letovicite), and (NH4)2SO4 (ammonium sulfate) can form. Once the sulfate has been neutralized, ammonium nitrate may form (Pandis and Seinfeld Citation2016); this is favored at lower temperatures and higher relative humidities (Krueger et al. Citation2003) and when gaseous nitric acid (HNO3) and ammonia (NH3) are abundant. Nitric acid can also react with sodium chloride from sea salt particles and calcium carbonate in the soil.
Many national parks and wilderness areas, collectively known as Class I areas, are protected from visibility degradation by the Clean Air Act and Regional Haze Rule (RHR) (U.S. EPA, Citation1999). Since 1985, the Interagency Monitoring of Protected Visual Environments (IMPROVE) network has collected extensive long-term monitoring data in these visually protected environments. Visibility degradation results from absorption and scattering of light by particle and gas-phase species in the air, typically expressed through a light extinction coefficient (bext). With considerable U.S. O/G development occurring close to national parks and other visually protected environments, there is concern that O/G activities could adversely affect air quality in these sensitive environments and hinder progress toward further reductions in regional haze.
Methods
Measurement site
The Carlsbad Caverns Air Quality Study (CarCavAQS) was conducted at CAVE in southeastern New Mexico from July 25 to September 5, 2019. Measurements were made at the CAVE Biology Office and building 58 (32.18° N, 104.44° W), located within the park, ~0.5 km from the park visitor center. The altitude of the site was approximately 1,355 m. Of the measurements discussed below, University Research Glassware (URG) denuder/filter pack inlets were set at approximately 1 meter above ground level (agl), and all other instrument inlets sampled from approximately 6-m agl. Meteorological data were collected at the same site location, using the meteorological station 35-015-0010, operated by the National Park Service (data can be found at: https://ard-request.air-resource.com/data.aspx).
Measurements
Measurements of meteorological conditions, VOCs, ozone, PM2.5, other gas species, and nitrogen deposition were collected at different sampling integration periods. Here, our focus is primarily on the characterization of aerosol mass and composition, along with concentrations of key trace gas species important either as aerosol precursors or as source indicators. The measurements used in this work and associated time resolutions are shown in . The IMPROVE sampler modules were installed shortly before the 2019 field campaign and continue to collect data for the network; all other instruments collected data only for the duration of this intensive.
Table 1. Measurements made during the CarCavAQS study are listed with their respective time-resolutions and measured species. Only instruments used in this work are listed. Abbreviations used for the measured species: sodium (Na+), ammonium (NH4+), calcium (Ca2+), potassium (K+), magnesium (Mg2+), chloride (Cl−), nitrite (NO2−), nitrate (NO3−), sulfate (SO42-), organic carbon (OC), black carbon (BC), elemental carbon (EC), ammonia (NH3), nitric acid (HNO3), sulfur dioxide (SO2).
The composition of water-soluble PM2.5 in near real-time was measured using Particle-into-Liquid samplers (PILS). A PILS-Ion Chromatography (PILS-IC) system measured inorganic cations and anions, and a PILS-Total Organic Carbon (PILS-TOC) system measured water-soluble organic carbon (WSOC) (Sullivan et al. Citation2006, Citation2004). The PILS mixes ambient particles with supersaturated water vapor to create droplets and inertially capture PM2.5 (Orsini et al. Citation2003; Weber et al. Citation2001). Each PILS sampled ambient air at 15 LPM with a 2.5 µm size-cut cyclone. Denuders coated with sodium carbonate and phosphoric acid were placed upstream of the PILS-IC to scrub out inorganic gases and an activated carbon parallel plate denuder (Eatough et al., Citation1993) was used to remove organic gases upstream of the PILS-TOC. For the PILS-TOC, to make a measurement of the background signal, a normally open actuated valve controlled by an external timer was periodically closed every 4 hours forcing the airflow through a Teflon filter before entering the PILS. The WSOC concentrations were calculated as the difference between the filtered and non-filtered measurements.
The PILS-IC system effluent was split into two Dionex ICS-1500 ICs for analysis of anions (Cl−, NO2−, NO3−, and SO42-) and cations (Na+, NH4+, K+, Ca2+, and Mg2+). Both systems utilized an isocratic pump, self-regenerating suppressor, and conductivity detector. Both ICs completed an analysis every 30 minutes, with a sample loop fill time of 13 minutes. The cations were separated using a Dionex IonPac CS12A analytical (4 × 250 mm) column with eluent of 18 mM methanesulfonic acid at a flow rate of 1.0 ml/min. A Dionex IonPac AS15 analytical (4 × 250 mm) column using an eluent of 38 mM sodium hydroxide at a flow rate of 1.5 ml/min was used for the anion analysis. The ICs were calibrated using authentic standards before the study, and a check standard was periodically injected during the study. The limit of detection for was 0.08 µg m−3 while for all other PILS-IC ions (Na+, NH4+, K+, Ca2+, Cl−, NO3−, and SO42-) it was 0.01 µg m−3.
In the PILS-TOC, to ensure insoluble particles were removed, the PILS liquid sample was pushed through a 0.2 µm PTFE liquid filter using syringe pumps. The liquid was then sent to a Sievers Model M9 Portable TOC Analyzer (Suez Waters Analytical Instruments, Boulder, CO). This instrument oxidizes organic carbon (OC) to CO2 using ammonium persulfate and ultraviolet light. Carbon dioxide formation is measured by conductivity. The amount of OC measured is proportional to the conductivity increase. The instrument was used in on-line mode with a 2-min integration time. The TOC Analyzer was calibrated in the factory and periodically verified by analysis of oxalic acid standards. The limit of detection for WSOC was 0.01 µg C m−3.
University Research Glassware (URG) annular denuder and filter pack samplers were used to measure inorganic gas and particle species (Allegrini et al. Citation1987, Citation1994; Fitz Citation2002). The air flow passed through a Teflon-coated PM2.5 cyclone and then through a sodium-carbonate-coated annular denuder to collect HNO3 (g) and SO2 (g), a phosphorous acid coated denuder to collect NH3 (g), and a 37 mm nylon filter (MTL Nylasorb, 1.0 μm pore size) to capture PM2.5 particles. Finally, an additional phosphorous acid coated denuder was located downstream of the filter to capture any NH3 volatilized from NH4NO3 initially captured on the filter. Any volatilized HNO3 is retained on the nylon filter (Yu et al. Citation2005). Full details are provided elsewhere (Benedict et al. Citation2013; Lee et al. Citation2008). The flow was controlled at 10 L min−1 and the pressure drop across the sample train was recorded. A dry gas meter downstream of the sample train measured the total flow integrated across the sampling period, which was then corrected for the pressure drop to obtain the ambient sample volume. URG samples were collected every 24 hours, from midnight to midnight. After sample collection, annular denuders and filters were extracted in 10 mL and 6 mL18.2 MΩ deionized water, respectively. Samples were then analyzed for anion and cation species using ion chromatography. Both systems utilized a Dionex DX-500 ion chromatograph (IC), which includes an isocratic pump, self-regenerating anion, or cation suppressor, and conductivity detector. The injection volume of both methods was 50 µL and analysis time was 17 minutes. The anion IC was used to quantify gas species ,
, and PM2.5 species Cl−, NO2−, NO3−, and SO42-. An IonPac AS14A (4 × 150 mm) analytical column was used with 1 mM sodium bicarbonate and 8 mM sodium bicarbonate eluent at a flow rate of 1 mL min−1. The cation IC was used to quantify NH3 (g), and PM2.5 species sodium (Na+), NH4+,
,
, and
. A Dionex IonPac CS12A (3x120 mm) analytical column was used with 20 mM methanesulfonic acid eluent at a flow rate of 0.5 mL min−1. Each component was quantified using an 8-standard calibration curve. A standard replicate and blank sample were analyzed after every 10 samples. Based on previous work, the relative standard deviations (RSDs) of major aerosol ion concentration measurements (NO3−, SO42-, and NH4+) are estimated to be between 3% and 5% and the RSDs for replicate denuder gas concentration measurements are estimated to be approximately 10% (Lee, Kreidenweis, and Collett Citation2004).
Each filter sample was also analyzed for levoglucosan using a Dionex DX-500 series ion chromatograph with detection via an ED-50/ED-50A electrochemical cell. This cell includes a gold working electrode and pH-Ag/AgCl (silver/silver chloride) reference electrode. A sodium hydroxide gradient and Dionex CarboPac PA-1 column (4 × 250 mm) were used. The complete run time was 59 min with an injection volume of 100 µL. More details can be found in Sullivan et al. (Citation2011a, Citation2011b, Citation2014, Citation2019). The limit of detection for levoglucosan was determined to be less than approximately 0.10 ng m−3.
IMPROVE monitoring sites include four sampling modules (A, B, C, and D) to measure particulate matter. The CAVE IMPROVE site began data collection on July 1, 2019, and data continue to be collected as a part of normal site operations (site code: CAVE1). Modules A, B, and C measure PM2.5 and provide the data sets considered in this work. Module A collects PM2.5 on a Teflon filter, which is then analyzed gravimetrically for PM2.5 mass and by X-ray fluorescence (XRF) for elemental composition (Prenni et al. Citation2016). Module B collects PM2.5 on a nylon filter, analyzed by IC for SO42-, NO3−, NO2−, and Cl−. Module C collects PM2.5 on a quartz filter that is analyzed by thermal optical reflectance (TOR) for OC and EC. (Chow et al. Citation2007; Malm et al. Citation2004). Further documentation can be found on the IMPROVE website (http://vista.cira.colostate.edu/Improve/).
A Magee-Scientific 7-channel Aethalometer (model AE31) measured PM2.5 light absorbing carbon at seven different wavelengths: 350, 470, 520, 590, 660, 880, and 950 nm (Hansen, Rosen, and Novakov Citation1984). Particles were collected on a quartz sample tape. An increase in light attenuation from the sampled location corresponds to an increase in collected light absorbing carbon. Absorption at 880 nm was used to determine the BC concentration (Drinovec et al. Citation2015). The aethalometer was operated using the standard procedure provided elsewhere (Petzold et al., Citation2013).
A Picarro gas analyzer (G2508) was used to quantify gas concentration of CH4, CO2, and NH3 every 15 seconds. CH4 concentrations were measured at parts per billion (ppb) sensitivity, with <7 ppb sensitivity at 1-min time resolution. All species are detected using cavity ring-down spectroscopy (CRDS). The Picarro sampled from a ¼” heated Teflon line, to limit the loss of NH3 (g), and through a fiber filter at its inlet, to remove particles. Before deployment, the calibration was verified by injection of known concentrations from certified cylinders. Zero measurements were made weekly by overflowing the inlet with ultra-high purity zero air for 30 min.
A Tapered Element Oscillating Microbalance (TEOM, model 1405-DF) from Thermo Scientific was used to measure the PM2.5 mass concentration (Clements et al. Citation2013). The TEOM 1405-DF uses diffusion drying rather than heating to better retain semi-volatile species. The TEOM measurement resolution is 0.1 µg m−3. The TEOM was calibrated in the field with a standard calibration filter from the manufacturer.
An Eco Physics NO (nitric oxide) analyzer coupled with converter inlets detected NO, NO2, and NOy, using NO-O3 chemiluminescence. NO2 was converted to NO by 395 nm photolysis (Pollack, Lerner, and Ryerson Citation2010) and NOy was converted to NO by a molybdenum converter heated to 320°C.
Reconstructing Visibility Impacts from Fine Particle Concentrations
The chemical composition of particulate matter, which is related to particle size distributions and determines the particle refractive index and hygroscopicity, is a key factor influencing light extinction. Potentially major contributors to total PM2.5 mass are (NH4)2SO4, NH4NO3, organic matter, light absorbing carbon (black or elemental carbon), soil dust, and sea salt (Malm Citation2016). Organic Matter (OM) is calculated as 1.8 times the organic carbon (Prenni et al. Citation2019). The second IMPROVE algorithm shown in EquationEquation (1)(1)
(1) calculates the light extinction (bext) based on fine particles, NO2, and Rayleigh Scattering. For this work, the light extinction contribution from coarse mass has not been considered. The second IMPROVE algorithm (EquationEquation 1
(1)
(1) ) divides some species into two different size bins (small and large) within the fine mode, to account for different light extinction properties based on particle size (Pitchford et al. Citation2007). The small and large bin sizes are determined by the total mass; a detailed explanation can be found in Prenni et al. (Citation2019). Hygroscopic species are multiplied by a growth factor, given as a function of relative humidity (RH). In EquationEquation (1)
(1)
(1) , fS (RH) and fL(RH) are pre-factors calculated as a function of RH for the small bin and large bin, respectively. Sea salt also has a corresponding pre-factor calculated as a function of RH, fSS (RH).
Fine Soil is calculated using EquationEquation (2)(2)
(2) , and sea salt is approximated by EquationEquation (3)
(3)
(3) using the chloride (Cl−) concentration (Malm et al. Citation2004). Soil is calculated as the sum of aluminum (Al), silicon (Si), titanium (Ti), calcium (Ca), and iron (Fe), multiplied by a respective constant.
More information on the specific variables can be found in Prenni et al. (Citation2019).
HYSPLIT back trajectories and residence time analysis
The National Oceanographic and Atmospheric Administration Air Resource Laboratory Hybrid Single-Particle Lagrangian Integrated Trajectory (HYSPLIT) model (Stein et al., Citation2015) was used to calculate back trajectories initiated from the CAVE monitoring site. Trajectories were generated for every hour of the study period. Trajectories were calculated using the NAM-12 reanalysis fields, which have a 12 km horizontal resolution, 26 atmospheric levels, and a 3-hr temporal resolution. The HYSPLIT vertical motion parameter was set to take vertical velocity fields directly from the meteorological model, the model top was set at 10,000 m-agl (meters above ground level), and the parcel was initiated at 10 m-agl. Only the first ensemble trajectory was used. HYSPLIT 24-hr trajectories and source–receptor relationships were investigated using Residence Time Analysis (RTA) (Gebhart et al. Citation2018, Citation2021). The residence time is the amount of time that a parcel spends in a horizontal grid-cell. This can be calculated as a probability, called the everyday probability (EP). Grid cells were defined with a 0.25° resolution. High concentration residence times or high probabilities (HP) are calculated using trajectories from the top 10% of concentration values at the receptor site. The top 10% of all concentration data points were verified to be greater than one standard deviation from the mean, to ensure they represented high values. These two residence times can be compared using the incremental probability (IP): the difference between HP and EP. The IP gives a comparison between HP and EP to determine if the probability of an endpoint being in a grid cell is more likely at a high concentration period probability or throughout the data set.
Results and discussion
Aerosol composition
To examine the impact of O/G and other sources on air quality in CAVE, various measurements are combined to look at the frequency and types of elevated PM2.5 events. Our analysis focuses on carbonaceous aerosol, inorganic ions, soil, and total PM2.5. The timelines of PM2.5 components measured at high time resolution are shown in , along with hourly average total PM2.5 mass. Episodic increases of many of the species are observed, but the duration and frequency of these events varies. Hourly averaged PM2.5 ranged up to 27 µg m−3 (maximum of 31.8 µg m−3 at 6-min resolution) and had an average value of 7.67 µg m−3. shows WSOC and BC, which had average concentrations of 1.2 µg C m−3 and 0.48 µg m−3, respectively.
Figure 2. Time series of major PM2.5 species and CH4 measured at the monitoring site. (a.) BC measured by the Aethalometer at 880 nm, WSOC measured by the PILS-TOC, and PM2.5 measured by the TEOM. Note that there is a gap in the PILS-IC and PILS-TOC data at the beginning of the study period associated with a power outage. The BC and WSOC are plotted at a time-resolution of 2-minutes. TEOM total PM2.5 is plotted at an hourly resolution. (b.) The five PILS-IC species with the largest contributions to total PM2.5 are plotted at 30-minute time-resolution. (c.) CH4 measured by the Picarro at 15-second time-resolution. Shaded regions represent case study transport dates.
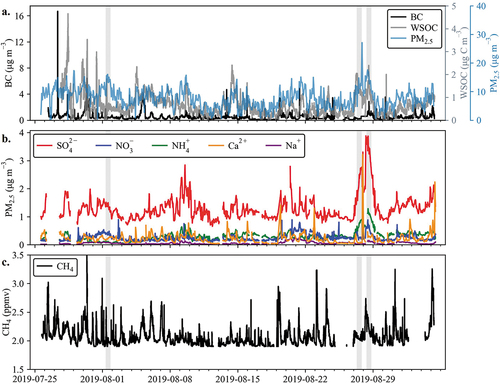
Several BC spikes exceeded 4 µg m−3. Although biomass burning can influence CAVE in summer, elevated BC measured in this campaign is likely not from biomass burning given the low concentrations of PM2.5 levoglucosan (avg. 2.2 ng m−3, ranging from 0.17 to 10.1 ng m−3), a good tracer for biomass burning (Sullivan et al. Citation2008), measured throughout the study. NO/NOx ratios and CH4 concentrations can be used to investigate possible sources. NOx is almost entirely emitted from combustion sources as NO; therefore, very fresh (minutes old) plumes present NO to NOx ratios near 1. None of the BC spikes correspond with NO/NOx ratios greater than 0.32 (study daytime avg. is 0.2), at 2-min time resolution, indicating that the elevated BC values do not come from very local sources, such as a diesel truck passing or idling near the sample inlet. CH4 is shown in and had an average mixing ratio of 2.1 ppmv (standard deviation of 0.16). The mean methane mixing ratio is 2.9 ppmv (standard deviation of 0.37) for all BC values above 4 µg m−3, suggesting that these high BC values could be associated with O/G flaring and/or diesel engine use. This will be further investigated using HYSPLIT back trajectory analysis later in this work.
shows timelines of the highest concentration species measured by the PILS-IC: SO42- (avg 1.3 µg m−3), NH4+, (avg 0.30 µg m−3), Ca2+ (avg 0.22 µg m−3), NO3− (avg 0.16 µg m−3), and Na+ (avg 0.057 µg m−3). The Ca2+ timeline exhibits several short-term, concentration spikes reaching as high as 3.2 µg m−3, suggesting emissions of soil dust from local erosion of limestone or gypsum deposits. There are significant gypsum (CaSO4) and dolomite (CaCO3) deposits in the Permian Basin (Johnson Citation1997), located near the CAVE monitoring site. Gypsum is a mineral made of Ca2+ and SO42- and could explain some of the excess SO42-, discussed later, and the presence of elevated Ca2+ concentrations. A significant proportion of the SO42- in White Sands National Park, another national park in the area, resulted from gypsum and other salts in the area during a study by White et al. (Citation2015). The presence of CaCO3 is challenging to confirm because carbonate cannot be measured directly by ion chromatography. The role of Ca2+ in the aerosol chemistry will be investigated further below.
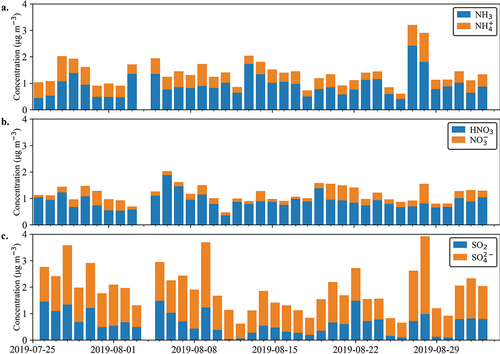
Concentrations of gas-phase NH3 (avg 0.93 µg m−3), HNO3 (avg 0.89 µg m−3), and SO2 (avg 0.64 µg m−3) and PM2.5 NH4+ (avg 0.47 µg m−3), NO3− (avg 0.27 µg m−3), and SO42+ (avg 1.3 µg m−3) measured with the 24-hr URG annular denuder/filter packs are shown in . The nitrogen species across nearly all days were dominated by the gas-phase components: NH3 and HNO3. Sulfur species were usually dominated by particle phase SO42-. The presence of significant gas-phase HNO3 and NH3 illustrates the potential for additional formation of particulate NO3−, either NH4NO3 formation under cooler more humid conditions, perhaps at other times of year, or additional coarse particle NO3− resulting from HNO3 reactive uptake on sodium chloride or soil dust particles.
Figure 3. URG annular denuder/filter pack sampling shows the particle/gas phase partitioning of (a.) NH3/NH4+, (b.) HNO3/ NO3−, and (c.) SO2/ SO42-. Particle species are shown in Orange, and gas species are shown in blue. There is one day of missing data, 2019–08-04, due to a denuder extraction error.
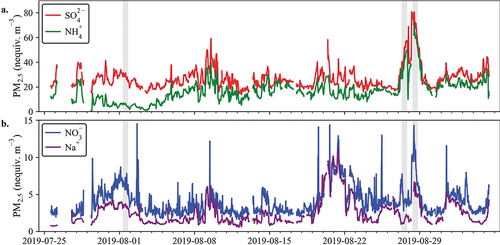
To better understand the aerosol molecular components and look at the pairing of aerosol ions, the concentrations were converted to charge equivalent units. Ions were paired based on likely combinations to form neutral aerosol. indicates that PM2.5 SO42- is only partially neutralized by NH4+, with an average NH4+ to SO42- charge equivalent ratio of 0.6. Twenty-four-hour NH3 (g) measured using the URG annular denuders averaged 0.93 µg m−3. In such a low humidity environment, it is expected that acidic SO42- aerosol (e.g., sulfuric acid or ammonium bisulfate) would take up NH3 (g) if available. The coexistence of NH3 (g) with SO42- not fully neutralized by NH4+ may indicate the presence of a different SO42- particle species.
Figure 4. PILS-IC species (a.) NH4+ and SO42-, and (b.) NO3− and Na+ are shown as nano equivalents m−3. Shaded regions represent case study transport dates.
Given that the study was conducted in summertime with high temperatures (avg. 28°C) and low RH (avg. 36%), the formation of significant NH4NO3 is not expected. HNO3 and its precursors can react with other species to form supermicron NO3− aerosols, such as sodium nitrate (NaNO3) or calcium nitrate (Ca(NO3)2), through reactions with sea salt and soil dust, respectively. Laboratory experiments support the formation of particle NO3− on sea salt and mineral dust particles from HNO3 and its precursors (Mamane and Gottlieb Citation1992). NaNO3 aerosol will form when sea salt (NaCl) reacts with nitric acid (HNO3) as shown in Reaction (1) (Pandis and Seinfeld Citation2016).
Comparison of the measured PM2.5 Cl− to Na+ ratios shows that they are consistently below the expected sea salt ratio of 1.16 (Lee et al. Citation2008), with an average ratio of 0.34 (R2 = 0.55), consistent with substantial hydrochloric acid displacement from the aerosol by reaction with nitric acid. A plot of this data can be found in the supplementary materials. Correlated concentration timelines of Na+ and NO3− (; R2 = 0.63) suggest that nitric acid displacement of chloride from sea salt may be important in this environment.
Reaction (2) shows HNO3 reaction with calcium carbonate (CaCO3) in soil dust which can lead to calcium nitrate aerosol formation.
CaCO3 and HNO3 readily react with each other at low RH values (Krueger et al. Citation2003) and this reaction has been found to be important for NO3− aerosol formation (Fairlie et al. Citation2010; Jordan et al. Citation2003). Reactions of nitric acid with sea salt and soil dust can compete and account for the formation of supermicron NO3− aerosol of differing composition.
During the Big Bend Regional Aerosol and Visibility Observational (BRAVO) in west Texas, researchers found that NO3− was mostly associated with sea salt particle Na+, but that the formation of Ca(NO3)2 was also important on several days (Lee, Kreidenweis, and Collett Citation2004). Malm et al. (Citation2005) found that in Yosemite National Park aerosol nitrate included both submicron NH4NO3 and substantial supermicron NaNO3 resulting from HNO3 reaction with sea salt transported inland across California’s Central Valley from the Pacific Ocean. Following these two studies, the coarse mode nitrate was characterized at several rural U.S. locations by Lee et al. (Citation2008). Of the five sites characterized, four had contributions from coarse-mode nitrate: Grand Canyon National Park, Great Smoky Mountains National Park, Brigantine National Wildlife Refuge, and San Gorgonio Wilderness Area. Springtime nitrate at the Southern California San Gorgonio mountain measuring site was mostly comprised of NH4NO3, while NaNO3 and Ca(NO3)2 together comprised approximately one-third of the total particle nitrate during measurements in the warmer summertime. Coarse mode nitrate formation from soil and sea salt accounted for the majority of particle nitrate in the Grand Canyon and the Great Smokey Mountains (Lee et al. Citation2008). Similar findings have also been reported outside the United States. Li and Shao (Citation2009) found the formation of Ca(NO3)2 to have a significant impact on brown haze episodes in northern China. Global simulations of the effects of mineral dust on aerosol nitrate found that nitrate concentrations in southern Europe, western U.S., and northeastern China are significantly impacted by mineral dust (Karydis et al. Citation2016). Across the globe, Karydis et al. found that the near surface fine mode nitrate increased by 21% when mineral dust reactions were included.
HNO3 (g) was present on all days during the 2019 CarCavAQS study period (see ). While much of the PM2.5 NO3− can be explained by a reaction between sea salt and HNO3, the fact that NO3− equivalent concentrations exceed Na+ equivalent concentrations suggests that an additional form of NO3− is also present. Reaction (2) is likely, given the elevated Ca2+ concentrations, availability of HNO3, and presence of limestone deposits in the area.
examines the ratio of cations and anions measured by the PILS-IC system more holistically. Ratios of Na+ plus NH4+ to SO42- plus NO3− always fall below unity, indicating an excess of anion species as discussed above. The presence of substantial NH3 (g) makes it unlikely that the aerosol is acidic enough for H+ to balance the excess anion levels. Adding Ca2+ to the (cation) numerator changes the story significantly. Currently many of the observations exhibit a cation/anion ratio close to 1, consistent with aerosol charge balance. There does appear to be a period of acidic aerosol from 07/30/2019 to 08/03/2019 and there are several shorter periods where the cation/anion ratio is above 1.0 and even exceeding 2.0. The vales where the ratio is the highest are associated with high Ca2+ concentrations. The deficiency in measured anion aerosol species during these periods likely represents the presence of considerable carbonate in this dust-rich aerosol (to be discussed further in section 3.2). As mentioned earlier, carbonate is not detected by ion chromatography. indicates that Ca2+ plays an important role in the PM2.5 aerosol balance, likely as either gypsum or calcium nitrate.
Figure 5. The ratio of cation to anion species measured by the PILS-IC is plotted in equiv. m−3. The dotted gray line shows a ratio of unity. The radius of the Orange point size changes proportionally with Ca2+ concentration. Shaded regions represent case study transport dates.
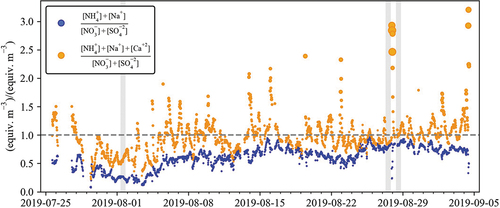
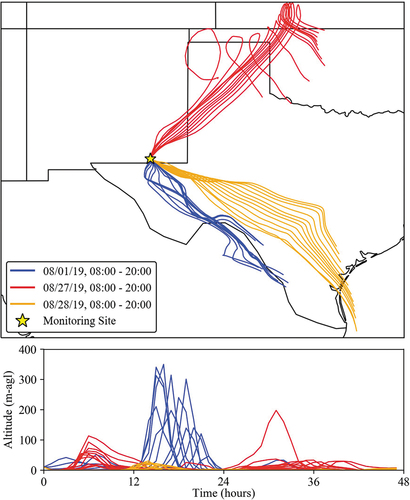
Transport during different aerosol composition periods was considered to help better understand how different source types and regions influence CAVE aerosol composition. HYSPLIT back-trajectories shown in , were run for 48 hours. A set of daytime (08:00 to 20:00) trajectories is shown for the period of acidic aerosol on 08/01/19 (see ), the period of highest gaseous NH3 on 08/27/19 (see ), and the largest total PM2.5 concentration from the PILS-IC on 08/28/19 (see ). Only the daytime trajectories are considered here due to increased variability in transport patterns overnight. Transport on 08/27/19 comes from the northeast and passes near two major coal-fired power plants shown in , which could be a source of the observed increase in sulfur compounds. This region also includes the largest agricultural production in the area based on the 2017 USDA Census of Agriculture (see map in the supplementary materials), consistent with elevated NH3 and NH4+ observed in the URG data on this date. Transport on 08/28/19 comes from the southeast over the largest concentration of O/G wells in the region and connects down to the Gulf of Mexico. This 48-hr transport pattern illustrates the importance of the Gulf of Mexico as a sea salt source region to support the reaction with HNO3 and the formation of NaNO3 discussed above. Measured aerosol on this date showed evidence of chloride depletion and correlation between PM2.5 Na+ and NO3−. The period of acidic aerosol on 08/01/19 also features transport from the southeast but shifted further south and moving more slowly than the 8/28/19 transport pattern. The air mass sampled at CAVE on this date has more limited NH3 from the URG denuder measurements and NH4+, consistent with the more acidic aerosol and the lack of major agricultural emissions in the upwind region. The gas-phase species CH4 and NOx also have distinct patterns across the 3 days considered, with both low on 08/01 and 08/27 and elevated on 08/28. The CH4 patterns can be seen clearly in the gray shaded regions of . Conversely, gaseous NH3 was low on 08/01 and 08/28 and elevated on 08/27. Low CH4 and NOx values on 08/01 are consistent with the sampled air masses passing to the south and west of the concentrated O/G region (see ). High CH4 and NOx mixing ratios on 08/28 correspond with a high period of NO3− and Na+, reflecting the importance of transport over the Permian O/G source region and upwind reactions between HNO3 and sea salt. NO3− and Na+ are consistently elevated from 08/20 through 08/22. The transport patterns for these days are consistent with 08/28 and have dominant transport from the Gulf of Mexico over the Permian O/G region. Overall transport patterns observed in the study will be considered in more detail in section 3.4.
Figure 6. 48-hour HYSPLIT Back Trajectories are shown for three different transport patterns during the study. The bottom panel shows back trajectory altitudes above ground level. The dates were selected based on aerosol composition. For each date 12 daytime back trajectories of 48-hour duration are plotted.
Measurement Comparisons and Mass Closure
PILS-TOC data averaged to daily frequency accounted for on average 78% of the IMPROVE OC. A value less than 100% is expected due to the presence of non-water-soluble OC components. Major species measured directly by IMPROVE and by the PILS (SO42-, NO3−, Ca2+, and Na+) agreed reasonably well, differing by less than 25% on average. Some difference is expected for calcium due to the differences in measuring water-soluble Ca2+ in the PILS-IC vs. elemental Ca in IMPROVE samples. IMPROVE Ca values were generally higher than PILS-IC Ca2+ values, consistent with expectations. Measured PM2.5 mass was compared between the IMPROVE and the TEOM data sets and differed by an average of 13%. A least squares linear regression, forced through the origin, between the paired values had a slope of 0.9 and a strong correlation (R2 = 0.79). Comparison figures between the aerosol mass and composition data sets can be found in the supplementary materials.
The sum of mass concentrations of the higher time resolution speciated PM2.5 measurements (PILS-IC species, BC, and WSOC multiplied by a factor of 1.8 to account for species beyond carbon in the organic aerosol) (Prenni et al. Citation2019) did not reach full mass closure when compared with the total PM2.5 mass measured by the TEOM. This is not surprising given the regional importance of insoluble or unquantified components tof airborne dust. The mean difference between the sum of the specified species and the total PM2.5 mass was 43%.
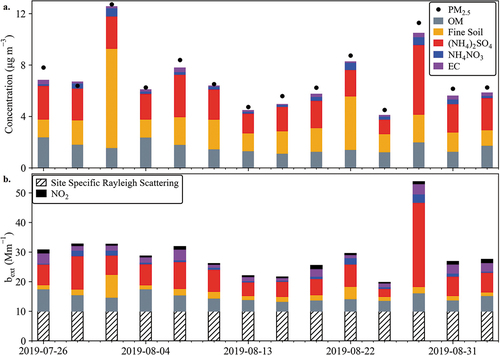
A timeline of the reconstructed PM2.5 mass from the IMPROVE measurements is plotted in . On average, the IMPROVE reconstructed mass represents 85% of the measured IMPROVE PM2.5 mass, ranging from 73% to 96%. The analysis of the IMPROVE filters includes elemental species by XRF, which captures the non-water-soluble mineral dust fraction of PM2.5 aerosol, as well as OC (vs. WSOC in the PILS). The reconstructed mass contains a significant contribution (average of 29.6% and maximum of 64.9%) from fine soil, calculated using EquationEquation 2(2)
(2) . This large contribution is consistent with the significant shortfall (average of 43%) in mass closure for the higher time resolution PILS aerosol measurements. IMPROVE mass reconstruction assumes that NO3− is present as NH4NO3 and the SO42- as (NH4)2SO4. Although the PILS-speciated data suggest that the NO3− is likely present as a mix of NaNO3 and Ca(NO3)2, this does not greatly impact the overall mass closure given the small amounts of NO3− measured and the similarity of the (charge) equivalent weights of Na+, Ca2+, and NH4+. The CAVE site-specific Rayleigh scattering is 10 Mm−1.
Figure 7. (a.) IMPROVE mass closure is plotted in the upper panel. Reconstructed PM2.5 mass is shown as the sum of OM, fine soil, (NH4)2SO4, NH4NO3, and EC. The IMPROVE measured PM2.5 mass is also shown by the black dots. (b.) The contribution to light extinction from each species plotted in (a.) as calculated using the second IMPROVE algorithm for light extinction. Site specific Rayleigh Scattering (10 Mm−1) and NO2 light extinction are also included. NO2 (g) light extinction is calculated using the second IMPROVE algorithm and a daily mean of the measured NO2.
Visibility impacts
Recent work by Hand et al. (Citation2020) looking at 1990–2018 trends across the United States indicates that haze formation in the Intermountain West/Southwest has decreased by ~1% per year, less than in many other regions. From 1990 to 2018, haze contributions by species have shifted to lower sulfate and more influence from carbonaceous aerosol and mineral dust. Average annual haze levels across the Intermountain West/Southwest region had an aerosol light extinction value similar to the background Rayleigh scattering value (10 Mm−1) (Hand et al. Citation2020). The 2019 measurements in CAVE are the first for this specific park.
shows the 24-hr light extinction values from each of the PM2.5 species (OM, fine soil, (NH4)2SO4, NH4NO3, and EC) measured by the CAVE IMPROVE filters. These values were calculated using the second IMPROVE algorithm (Pitchford et al. Citation2007; Prenni et al. Citation2019). This light extinction calculation assumes that NO3− is present as NH4NO3. Light extinction is based on refractive index, assumed size distribution, and hygroscopicity, which differ for NH4NO3 vs. NaNO3 or Ca(NO3)2. In the end, however, the 24-hr light extinction from NO3− is a small contributor relative to other species.
The reconstructed PM2.5 light extinction for IMPROVE sample days is typically dominated by SO42-, with a mean fractional contribution of 43% and a range from 28% to 66% during the study. Extinction by organic matter, EC, fine soil, and NH4NO3 average 27%, 12%, 12%, and 5.3%, respectively. OM was the largest contributor to particle light extinction on 3 days (July 26, August 4, and 25). Fine soil was the largest contributor on August 1, 2019. Although SO42- generally dominates the particle light extinction, the presence of carbonaceous aerosol and mineral dust as the largest contributors on given days, is consistent with the more regional findings of Hand et al. (Citation2020) discussed above.
To better understand the short-term variability in light extinction and associated aerosol species’ contributions, the second IMPROVE algorithm was also applied to our higher time resolution data sets. To do this several assumptions were needed. Some of these assumptions impact the absolute accuracy of the estimated extinction, but the goal is to ascertain relative variability in light extinction and whether short-term concentration excursions lead to species other than SO42-, OM, or fine soil dominating the extinction budget over periods shorter than the 24-hr IMPROVE sample periods. SO42- and NO3− were again assumed to be present as (NH4)2SO4 and NH4NO3, consistent with the IMPROVE light extinction calculations above. Aethalometer BC (880 nm) was assumed to be a good proxy for EC. To estimate fine soil, study average IMPROVE concentrations were used for each elemental species in EquationEquation 2(2)
(2) , except for calcium. Calcium exhibited the largest range of fractional contribution to IMPROVE fine soil by a significant margin and was the only species in the IMPROVE soil equation measured in the PILS. The PILS, however, measured water-soluble Ca2+ ion, which represents a fraction of the elemental Ca present and measured by XRF in the IMPROVE samples. A comparison of PILS Ca2+ vs. IMPROVE Ca revealed that water-soluble Ca2+ compromised an average of 78% of elemental Ca, with a range of 48% to 100%, indicating that our substitution of Ca2+ for Ca in the IMPROVE soil equation represents a lower bound on that component of fine soil. PILS WSOC was used as a surrogate for OC, so we are again estimating a lower bound for the OC contribution to light extinction. The IMPROVE scale factor (1.8) was used to convert WSOC to OM.
Utilizing these assumptions, averaging high time resolution concentration measurements to 1 hr, and making use of hourly meteorological data from the National Park Service at CAVE, light extinction was calculated at hourly time frequency (). The maximum bext values, likely lower bounds as discussed above, reach 90 Mm−1 vs. a daily maximum of 54 Mm−1 determined from the IMPROVE measurements.
Figure 8. The second IMPROVE equation was applied to higher time resolution composition data sets (PILS-IC, PILS-TOC, and Aethalometer BC) to estimate hourly light extinction values. These values include the Rayleigh background contribution as well as extinction from NO2. Fractional light extinction contributions, excluding Rayleigh background, from different species are shown for three data points (shown as blue triangles) as examples. Extinction peaks highlighting maximum contributions are represented by points a and c (30 July 2019 09:00 MST and 28 August 2019 12:00 MST). Point b represents a low light extinction period on 18 August 2019 12:00 MST.
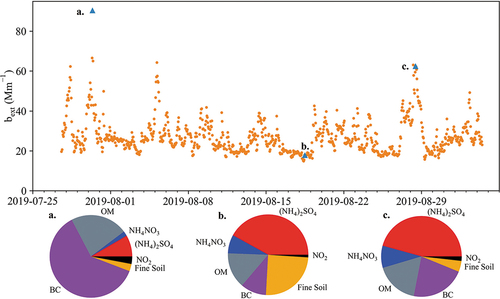
The bext peaks represented by points “a” and “c” in highlight maximum contributions to light extinction by different species. Point “a” has a majority contribution from BC. As discussed earlier, the NO/NOx ratio at this time is low (ratio of 0.22) and similar to other time periods (daytime avg 0.2), likely ruling out a very local combustion source (e.g., an idling vehicle near the inlet), as does the duration of this extinction event across multiple hours. A high CH4 mixing ratio of 3.16 ppmv (area background of 2 ppmv) coinciding with this light extinction peak suggests O/G activity, likely flaring or diesel engine emissions, as a source of the BC that dominates this 90 Mm−1 hourly extinction episode. Both points “b” and “c”, with low and high hourly bext respectively, have the largest extinction contributions from SO42-, similar to the typical pattern at daily time resolution using the IMPROVE data. Both high extinction points (a and c) have corresponding NOAA HYSPLIT back-trajectories that indicate transport from the southeast, a region of intense O/G development and production. The high variability in hourly light extinction values and the different aerosol compositions during peak extinction episodes point to the importance of high time resolution measurements to adequately characterize peaks in visibility impairment in the park and the activities responsible for those episodes.
Residence time analysis
Incremental probability (IP) distributions for the residence time analyses conducted for major species and associated light extinction are presented in . Red grid cells indicate that an air mass was more likely to have passed through that grid cell on a high concentration/extinction trajectory (top 10% of parameter values) than during the full set of study back-trajectories. Blue grid cells indicate that passage of a trajectory through that cell was more likely during the full trajectory set than on a high concentration/extinction trajectory.
Figure 9. Incremental probability distributions are plotted for 7 species measured directly during the field campaign: SO42-, NH4+, WSOC, Ca2+, NO3−, BC, and CH4 and 1 calculated value: particle light extinction (bext). The IPs are based on 24-hr back trajectories and are distance normalized. This is done by multiplying each grid cell value by the distance from the origin site, shown as a black dot. The grid cell resolution is 0.25 degrees square.
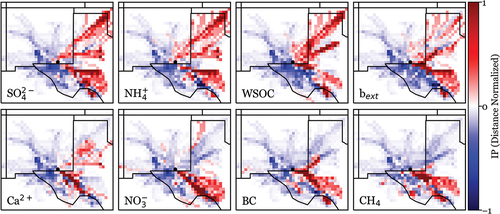
High concentration periods of SO42-, NH4+, and WSOC show prevailing directionality from the SE and from the NE. The high concentration trajectory similarity for SO42- and NH4+ reflects the tight association of aerosol NH4+ with SO42-. As discussed above, coal-fired power plants and agricultural activity to the NE may contribute to increased SO42- and NH3/NH4+ concentrations during this transport pattern. The light extinction plot (bext) shows that the most haze-impacted periods are more strongly associated with transport from the SE, although some influence from the NE remains. The bottom row of species in the figure (Ca2+, NO3−, BC, and CH4) shows the highest concentration measurement periods strongly associated with transport from the SE. The SE sector corresponds with the highest concentration of nearby O/G activity, a likely source of NO3− precursor NOx, BC, and CH4. The SE sector also corresponds with Permian Basin limestone deposits, a likely source of soil dust rich in calcium carbonate. Transport from this direction can also bring sea salt from the Gulf of Mexico, as discussed above, providing an additional path to generate aerosol NO3− via HNO3 uptake on sea salt.
Summary
The CarCavAQS study in Carlsbad Caverns National Park provided novel and important insight into the composition and likely sources of particulate matter in the region. PM2.5 mass ranged up to 31.8 µg m−3 with major inorganic contributions from SO42-, NH4+, NO3−, Na+, and Ca2+. BC and WSOC ranged up to 16.7 µg m−3 and 4.7 µg C m−3, respectively. Fine particle SO42- is partially neutralized by NH4+, but likely also includes a component from gypsum (CaSO4) found in regional soils. Summertime NO3− appears to be present as a result of abundant HNO3 or its precursors reacting with NaCl from sea salt and CaCO3 from mineral dust. The gas phases NH3 and HNO3 exceed their particle-phase counterparts throughout the study. Forty-eight-hour back trajectory analysis of three distinct transport patterns indicates that air masses often pass through regions with substantial O/G activity when transported to the site from the SE. Transport from this direction often originates over the Gulf of Mexico, carrying sea salt which can react with O/G emissions such as HNO3 or its precursors. Episodes of high BC concentrations appear to be associated with O/G activity. Aerosol light extinction showed considerable variability at hourly timescales, reaching a maximum of 90 Mm−1 during the study. This temporal variability illustrates the importance of high time resolution measurements for understanding peaks in regional haze at protected locations. While SO42-, on average, was the largest fine particle contributor to light extinction, BC, likely emitted by the abundant flaring or other Permian Basin O/G activities, was found to dominate one of the haziest periods. The highest concentration/light extinction air masses were transported predominantly from the SE, a region with a high density of Permian Basin O/G operations. Future anticipated increases in O/G extraction in the area could exacerbate the air quality problems documented during this study.
Supplementary_Materials_Naimie_et_al_CAVE.docx
Download MS Word (1.4 MB)Acknowledgment
The authors thank officials at Carlsbad Caverns National Park for providing study access and logistical support. This work was supported by the National Park Service under Agreement Number P20AC00679 with Colorado State University.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are available from the corresponding author, Jeffrey L. Collett, upon reasonable request. https://doi.org/10.25675/10217/235481.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/10962247.2022.2081634
Additional information
Funding
Notes on contributors
Lillian E. Naimie
Lillian E. Naimie received a B.A. in Chemistry from Colby College. She is currently a Ph.D. student at Colorado State University in Atmospheric Science.
Amy P. Sullivan
Amy P. Sullivan is a Research Scientist III in Atmospheric Science at Colorado State University.
K.B. Benedict
K.B. Benedict is a Research Scientist at Los Alamos National Laboratory. During the data collection of this work, she was a Research Scientist in Atmospheric Science at Colorado State University.
Anthony J. Prenni
Anthony J. Prenni is a chemist in the Air Resources Division of the National Park Service.
B.C. Sive
B.C. Sive is an atmospheric chemist in the Air Resources Division of the National Park Service where he serves as the Program Manager for the Gaseous Pollutant Monitoring Program (GPMP).
Bret A. Schichtel
Bret A. Schichtel is a physical scientist in the Air Resource Division of the National Park Service and an affiliate Research Scientist at the CSU-Cooperative Institute for Research in the Atmosphere.
Emily V. Fischer
Emily V. Fischer is an Associate Professor of Atmospheric Science at Colorado State University.
Ilana Pollack
Ilana Pollack is a Research Scientist III in Atmospheric Science at Colorado State University.
Jeffrey Collett
Jeffrey Collett, Jr. is a Professor and Department Head for Atmospheric Science at Colorado State University.
References
- Allegrini, I., F. De Santis, V. Di Palo, A. Febo, C. Perrino, M. Possanzini, and A. Liberti. 1987. Annular denuder method for sampling reactive gases and aerosols in the atmosphere. Sci. Total Environ. 67 (1):1–16. doi:10.1016/0048-9697(87)90062-3.
- Allegrini, I., A. Febo, C. Perrino, and P. Masia. 1994. Measurement of atmospheric nitric acid in gas phase and nitrate in particulate matter by means of annular denuders. Int. J. Environ. Anal. Chem. 54 (3):183–201. doi:10.1080/03067319408034088.
- Allen, D. T., D. Smith, V. M. Torres, and F. C. Saldaña. 2016. Carbon dioxide, methane and black carbon emissions from upstream oil and gas flaring in the United States. Curr. Opin. Chem. Eng. 13:119–23. doi:10.1016/j.coche.2016.08.014.
- Benedict, K. B., C. M. Carrico, S. M. Kreidenweis, B. Schichtel, W. C. Malm, and J. L. Collett. 2013. A seasonal nitrogen deposition budget for Rocky Mountain National Park. Ecol. Appl. 23 (5):1156–69. doi:10.1890/12-1624.1.
- Benedict, K. B., A. J. Prenni, M. M. H. El-Sayed, A. Hecobian, Y. Zhou, K. A. Gebhart, B. C. Sive, B. A. Schichtel, and J. L. Collett. 2020. Volatile organic compounds and ozone at four national parks in the southwestern United States. Atmos. Environ. 239:117783. doi:10.1016/j.atmosenv.2020.117783.
- Bond, T. C., B. Wehner, A. Plewka, A. Wiedensohler, J. Heintzenberg, and R. J. Charlson. 2006. Climate-relevant properties of primary particulate emissions from oil and natural gas combustion. Atmos. Environ. 40 (19):3574–87. doi:10.1016/j.atmosenv.2005.12.030.
- Bond, T. C., S. J. Doherty, D. W. Fahey, P. M. Forster, T. Berntsen, B. J. DeAngelo, M. G. Flanner, S. Ghan, B. Kärcher, D. Koch, et al. 2013. Bounding the role of black carbon in the climate system: A scientific assessment. J. Geophys. Res. Atmos. 118 (11):5380–552. doi:10.1002/jgrd.50171.
- Böttcher, K., -V.-V. Paunu, K. Kupiainen, M. Zhizhin, A. Matveev, M. Savolahti, Z. Klimont, S. Väätäinen, H. Lamberg, and N. Karvosenoja. 2021. Black carbon emissions from flaring in Russia in the period 2012–2017. Atmos. Environ. 254:118390. doi:10.1016/j.atmosenv.2021.118390.
- Brown, D., C. Lewis, and B. Weinberger. 2015. Human exposure to unconventional natural gas development: A public health demonstration of periodic high exposure to chemical mixtures in ambient air. J. Environ. Sci. Health. A Tox. Hazard. Subst. Environ. Eng. 50:460–72. doi:10.1080/10934529.2015.992663.
- Chow, J. C., J. G. Watson, L. W. A. Chen, M. C. O. Chang, N. F. Robinson, D. Trimble, and S. Kohl. 2007. The IMPROVE_A temperature protocol for thermal/optical carbon analysis: Maintaining consistency with a long-term database. J. Air Waste Manage. Assoc. 57 (9):1014–23. doi:10.3155/1047-3289.57.9.1014.
- Clements, N., J. B. Milford, S. L. Miller, W. Navidi, J. L. Peel, and M. P. Hannigan. 2013. Errors in coarse particulate matter mass concentrations and spatiotemporal characteristics when using subtraction estimation methods. J. Air Waste Manage. Assoc. 63 (12):1386–98. doi:10.1080/10962247.2013.816643.
- Conrad, B. M., and M. R. Johnson. 2019. Mass absorption cross-section of flare-generated black carbon: Variability, predictive model, and implications. Carbon 149:760–71. doi:10.1016/j.carbon.2019.04.086.
- Drinovec, L., G. Močnik, P. Zotter, A. S. H. Prévôt, C. Ruckstuhl, E. Coz, M. Rupakheti, J. Sciare, T. Müller, A. Wiedensohler, et al. 2015. The ”dual-spot” Aethalometer: An improved measurement of aerosol black carbon with real-time loading compensation. Atmos. Meas. Tech. 8 (5):1965–79. doi:10.5194/amt-8-1965-2015.
- Eatough, D. J., A. Wadsworth, D. A. Eatough, J. W. Crawford, L. D. Hansen, and E. A. Lewis. 1993. A multiple-system, multi-channel diffusion denuder sampler for the determination of fine-particulate organic material in the atmosphere. Atmospheric Environment. Part A. General Topics 27 (8):1213–19. doi:10.1016/0960-1686(93)90247-V.
- Elvidge, C. D., D. Ziskin, K. E. Baugh, B. T. Tuttle, T. Ghosh, D. W. Pack, E. H. Erwin, and M. Zhizhin. 2009. A fifteen year record of global natural gas flaring derived from satellite data. Energies 2 (3):595–622. doi:10.3390/en20300595.
- Evanoski-Cole, A. R., K. A. Gebhart, B. C. Sive, Y. Zhou, S. L. Capps, D. E. Day, A. J. Prenni, M. I. Schurman, A. P. Sullivan, Y. Li, et al. 2017. Composition and sources of winter haze in the Bakken oil and gas extraction region. Atmos. Environ. 156:77–87. doi:10.1016/j.atmosenv.2017.02.019.
- Fairlie, T. D., D. J. Jacob, J. E. Dibb, B. Alexander, M. A. Avery, A. van Donkelaar, and L. Zhang. 2010. Impact of mineral dust on nitrate, sulfate, and ozone in transpacific Asian pollution plumes. Atmos. Chem. Phys 10 (8):3999–4012. doi:10.5194/acp-10-3999-2010.
- Field, R. A., J. Soltis, M. C. McCarthy, S. Murphy, and D. C. Montague. 2015. Influence of oil and gas field operations on spatial and temporal distributions of atmospheric non-methane hydrocarbons and their effect on ozone formation in winter. Atmos. Chem. Phys 15 (6):3527–42. doi:10.5194/acp-15-3527-2015.
- Fitz, D. R. 2002. Evaluation of Diffusion Denuder Coatings for Removing Acid Gases from Ambient AirRep. University of California, Riverside: Office of Air Quality Planning and Standards, U.S. Environmental Protection Agency.
- Gaswirth, S. B., K. L. French, J. K. Pitman, K. R. Marra, T. J. Mercier, H. M. Leathers-Miller, C. J. Schenk, M. E. Tennyson, C. A. Woodall, and M. E. Brownfield, et al. 2018. Assessment of undiscovered continuous oil and gas resources in the Wolfcamp Shale and Bone Spring Formation of the Delaware Basin, Permian Basin Province, New Mexico and Texas, 2018. Report Rep. 2018-3073, Reston, VA. https://doi.org/10.3133/fs20183073
- Gebhart, K. A., D. E. Day, A. J. Prenni, B. A. Schichtel, J. L. Hand, and A. R. Evanoski-Cole. 2018. Visibility impacts at Class I areas near the Bakken oil and gas development. J. Air Waste Manage. Assoc. 68 (5):477–93. doi:10.1080/10962247.2018.1429334.
- Gebhart, K., R. Farber, J. Hand, D. Eatough, B. Schichtel, J. Vimont, M. Green, and W. Malm. 2021. Long-Term trends in particulate sulfate at grand canyon national park. Atmos. Environ. 253:118339. doi:10.1016/j.atmosenv.2021.118339.
- Hand, J. L., A. J. Prenni, S. Copeland, B. A. Schichtel, and W. C. Malm. 2020. Thirty years of the Clean Air Act Amendments: Impacts on haze in remote regions of the United States (1990–2018. Atmos. Environ. 243:117865. doi:10.1016/j.atmosenv.2020.117865.
- Hansen, A. D. A., H. Rosen, and T. Novakov. 1984. The aethalometer — An instrument for the real-time measurement of optical absorption by aerosol particles. Sci. Total Environ. 36 (36):191–96. doi:10.1016/0048-9697(84)90265-1.
- Hecobian, A., A. L. Clements, K. B. Shonkwiler, Y. Zhou, L. P. MacDonald, N. Hilliard, B. L. Wells, B. Bibeau, J. M. Ham, J. R. Pierce, et al. 2019. Air toxics and other volatile organic compound emissions from unconventional oil and gas development. Environ. Sci. Technol. Lett. 6 (12):720–26. doi:10.1021/acs.estlett.9b00591.
- Homeland Infastructure Foundation-Level Data (HIFLD). 2019. Oil and natural gas wells. Accessed February 26, 2021. https://gii.dhs.gov/HIFLD.
- Johnson, K. S. 1997. Permian evaporites in the Permian Basin of southwestern United States. Pr. Państw. Inst. Geol. (157 PART 2):95–96.
- Jordan, C., J. Dibb, B. Anderson, M. H. Hecht, S. M. Grannan-Feldman, K. Manatt, S. J. West, X. Wen, M. Frant, and T. Gillette. 2003. Uptake of nitrate and sulfate on dust aerosols during TRACE-P. J. Geophys. Res 108:13-1 - 13–12. doi:10.1029/2002JD003101.
- Karydis, V. A., A. P. Tsimpidi, A. Pozzer, M. Astitha, and J. Lelieveld. 2016. Effects of mineral dust on global atmospheric nitrate concentrations. Atmos. Chem. Phys 16:1491–509. doi:10.5194/acp-16-1491-2016.
- Khalek, I. A., M. G. Blanks, P. M. Merritt, and B. Zielinska. 2015. Regulated and unregulated emissions from modern 2010 emissions-compliant heavy-duty on-highway diesel engines. J. Air Waste Manage. Assoc. 65 (8):987–1001. doi:10.1080/10962247.2015.1051606.
- Krueger, B. J., V. H. Grassian, A. Laskin, and J. P. Cowin. 2003. The transformation of solid atmospheric particles into liquid droplets through heterogeneous chemistry: Laboratory insights into the processing of calcium containing mineral dust aerosol in the troposphere. Geophys. Res. Lett. 30 (3). doi: 10.1029/2002GL016563.
- Lee, T., S. M. Kreidenweis, and J. L. Collett. 2004. Aerosol ion characteristics during the big bend regional aerosol and visibility observational study. J. Air Waste Manage. Assoc. 54 (5):585–92. doi:10.1080/10473289.2004.10470927.
- Lee, T., X.-Y. Yu, B. Ayres, S. M. Kreidenweis, W. C. Malm, and J. L. Collettjr. 2008. Observations of fine and coarse particle nitrate at several rural locations in the United States. Atmos. Environ. 42 (11):2720–32. doi:10.1016/j.atmosenv.2007.05.016.
- Li, W. J., and L. Y. Shao. 2009. Observation of nitrate coatings on atmospheric mineral dust particles. Atmos. Chem. Phys 9:1863–71. doi:10.5194/acp-9-1863-2009.
- Li, Y., B. A. Schichtel, J. T. Walker, D. B. Schwede, X. Chen, C. M. Lehmann, M. A. Puchalski, D. A. Gay, and J. L. Collett. 2012. Increasing importance of deposition of reduced nitrogen in the United States. Proc. Natl. Acad. Sci. 113:5874– 79. doi:10.1073/pnas.1525736113
- Malm, W. C., B. A. Schichtel, M. L. Pitchford, L. L. Ashbaugh, and R. A. Eldred. 2004. Spatial and monthly trends in speciated fine particle concentration in the United States. J. Geophys. Res. Atmos. 109 (D3). doi: 10.1029/2003JD003739.
- Malm, W. C., D. E. Day, C. Carrico, S. M. Kreidenweis, J. L. Collett, G. McMeeking, T. Lee, J. Carrillo, and B. Schichtel. 2005. Intercomparison and closure calculations using measurements of aerosol species and optical properties during the Yosemite Aerosol Characterization Study. J. Geophys. Res 110:D14302. doi:10.1029/2004JD005494.
- Malm, W. C. 2016. Chapter 1 - Introduction. In Visibility, 1–27. Elsevier. doi:10.1016/B978-0-12-804450-6.00001-2.
- Mamane, Y., and J. Gottlieb. 1992. Nitrate formation on sea-salt and mineral particles—a single particle approach. Atmos. Environ. A, Gen. Top. 26 (9):1763–69. doi:10.1016/0960-1686(92)90073-T.
- Olaguer, E. P. 2012. The potential near-source ozone impacts of upstream oil and gas industry emissions. J. Air Waste Manage. Assoc. 62 (8):966–77. doi:10.1080/10962247.2012.688923.
- Orsini, D., Y. Ma, A. Sullivan, B. Sierau, K. Baumann, and R. Weber. 2003. Refinements to the particle-into-liquid sampler (PILS) for ground and airborne measurements of water-soluble aerosol composition. Atmos. Environ. 37:1243–59. doi:10.1016/S1352-2310(02)01015-4.
- Pandis, S. N., and J. H. Seinfeld. 2016. Atmospheric chemistry and physics: From air pollution to climate change. Hoboken, NJ: Wiley-Blackwell.
- Petzold, A., J. A. Ogren, M. Fiebig, P. Laj, S. M. Li, U. Baltensperger, T. Holzer-Popp, S. Kinne, G. Pappalardo, N. Sugimoto, et al. 2013. Recommendations for the interpretation of “black carbon” measurements. Atmospheric Chem. Phys. 13. doi:10.5194/acpd-13-9485-2013.
- Pitchford, M., W. Malm, B. Schichtel, N. Kumar, D. Lowenthal, and J. Hand. 2007. Revised Algorithm for Estimating Light Extinction from IMPROVE Particle Speciation Data. J. Air Waste Manage. Assoc. 57 (11):1326–36. doi:10.3155/1047-3289.57.11.1326.
- Pollack, I. B., B. M. Lerner, and T. B. Ryerson. 2010. Evaluation of ultraviolet light-emitting diodes for detection of atmospheric NO2 by photolysis - chemiluminescence. J. Atmos. Chem. 65 (2):111–25. doi:10.1007/s10874-011-9184-3.
- Prenni, A. J., D. E. Day, A. R. Evanoski-Cole, B. C. Sive, A. Hecobian, Y. Zhou, K. A. Gebhart, J. L. Hand, A. P. Sullivan, Y. Li, et al. 2016. Oil and gas impacts on air quality in federal lands in the Bakken region: An overview of the Bakken Air Quality Study and first results. Atmos. Chem. Phys 16 (3):1401–16. doi:10.5194/acp-16-1401-2016.
- Prenni, A., J. Hand, W. Malm, S. Copeland, G. Luo, F. Yu, N. Taylor, L. Russell, and B. Schichtel. 2019. An examination of the algorithm for estimating light extinction from IMPROVE particle speciation data. Atmos. Environ. 214:116880. doi:10.1016/j.atmosenv.2019.116880.
- Shen, X., T. Lv, X. Zhang, X. Cao, X. Li, B. Wu, X. Yao, Y. Shi, Q. Zhou, X. Chen, et al. 2021. Real-world emission characteristics of black carbon emitted by on-road China IV and China V diesel trucks. Sci. Total Environ. 799:149435. doi:10.1016/j.scitotenv.2021.149435.
- Stein, A. F., R. R. Draxler, G. D. Rolph, B. J. B. Stunder, M. D. Cohen, and F. Ngan. 2015. NOAA’s HYSPLIT Atmospheric Transport and Dispersion Modeling System. Bull. Am. Meteorol. Soc. 96 (12):2059–77. doi:10.1175/BAMS-D-14-00110.1.
- Sullivan, A., R. Weber, A. Clements, J. Turner, M. Bae, and J. Schauer. 2004. A method for on-line measurement of water-soluble organic carbon in ambient aerosol particles: Results from an urban site. Geophys. Res. Lett. 311. doi:10.1029/2004GL019681.
- Sullivan, A. P., R. E. Peltier, C. A. Brock, J. A. de Gouw, J. S. Holloway, C. Warneke, A. G. Wollny, and R. J. Weber. 2006. Airborne measurements of carbonaceous aerosol soluble in water over northeastern United States: Method development and an investigation into water-soluble organic carbon sources. J. Geophys. Res. Atmos. 111 (D23). doi: 10.1029/2006JD007072.
- Sullivan, A. P., A. S. Holden, L. A. Patterson, G. R. McMeeking, S. M. Kreidenweis, W. C. Malm, W. M. Hao, C. E. Wold, and J. L. Collett Jr. 2008. A method for smoke marker measurements and its potential application for determining the contribution of biomass burning from wildfires and prescribed fires to ambient PM2.5 Organic Carbon. J. Geophys. Res 113:D22302. doi:10.1029/2008JD010216.
- Sullivan, A. P., N. Frank, G. Onstad, C. D. Simpson, and J. L. Collett Jr. 2011a. Application of High-Performance Anion-Exchange Chromatography – Pulsed Amperometric Detection for Measuring Carbohydrates in Routine Daily Filter Samples Collected by a National Network: 1. Determination of the Impact of Biomass Burning in the Upper Midwest. J. Geophys. Res 116:D08302. doi:10.1029/2010JD014166.
- Sullivan, A. P., N. Frank, D. M. Kenski, and J. L. Collett Jr. 2011b. Application of high-Performance Anion-Exchange Chromatography – Pulsed amperometric detection for measuring carbohydrates in routine daily filter samples collected by a national network: 2. examination of sugar alcohols/polyols, sugars, and anhydrosugars in the Upper Midwest. J. Geophys. Res 116:D08303. doi:10.1029/2010JD014169.
- Sullivan, A. P., A. A. May, T. Lee, G. R. McMeeking, S. M. Kreidenweis, S. K. Akagi, R. J. Yokelson, S. P. Urbanski, and J. L. Collett Jr. 2014. Airborne-Based source smoke marker ratios from prescribed burning. Atmos. Chem. Phys 14:10535–45. doi:10.5194/acp-14-10535-2014.
- Sullivan, A. P., H. Guo, J. C. Schroder, P. Campuzano-Jost, J. L. Jimenez, T. Campos, V. Shah, L. Jaeglé, B. H. Lee, F. D. Lopez‐hilfiker, et al. 2019. Biomass burning markers and residential burning in the WINTER aircraft campaign. Journal of Geophysical Research: Atmospheres 124:1846–61. doi:10.1029/2017JD028153.
- Thompson, C. R., J. Hueber, and D. Helmig. 2014. Influence of oil and gas emissions on ambient atmospheric non-methane hydrocarbons in residential areas of Northeastern Colorado. Elementa 3. doi:10.12952/journal.elementa.000035.
- United States Energy Information Administration (EIA). 2021a. Where our natural gas comes from. Accessed November 11, 2021. https://www.eia.gov/energyexplained/natural-gas/where-our-natural-gas-comes-from.php.
- United States Energy Information Administration (EIA). 2021b. Annual Energy Outlook. Accessed November 10, 2021, from https://www.eia.gov/outlooks/aeo/.
- United States Energy Information Administration (EIA). 2022. U.S. Energy mapping system. Accessed March 21, 2022. https://www.eia.gov/state/maps.php.
- United States Environmental Protection Agency (EPA). 1999. Regional haze regulations: Final rule. 40 CFR Part 51. Fed. Regist. 64: 35714–74.
- United States Environmental Protection Agency (EPA). 2019. 2017 national emissions inventory (NEI) data. Environmental Protection Agency. Accessed April 8, 2022, from https://www.epa.gov/air-emissions-inventories/2017-national-emissionsinventory-nei-data
- USDA National Agricultural Statistics Service. 2017. Census of agriculture. www.nass.usda.gov/AgCensus.
- Weber, R. J., D. Orsini, Y. Daun, Y. N. Lee, P. J. Klotz, and F. Brechtel. 2001. A Particle-into-Liquid Collector for Rapid Measurement of Aerosol Bulk Chemical Composition. Aerosol Sci. Technol. 35 (3):718–27. doi:10.1080/02786820152546761.
- Weyant, C. L., P. B. Shepson, R. Subramanian, M. O. L. Cambaliza, A. Heimburger, D. McCabe, E. Baum, B. H. Stirm, and T. C. Bond. 2016. Black carbon emissions from associated natural gas flaring. Environ. Sci. Technol. 50 (4):2075–81. doi:10.1021/acs.est.5b04712.
- White, W. H., N. P. Hyslop, K. Trzepla, S. Yatkin, R. S. Rarig Jr, T. E. Gill, and L. Jin. 2015. Regional transport of a chemically distinctive dust: Gypsum from White Sands, New Mexico (USA. Aeolian Res. 16:1–10. doi:10.1016/j.aeolia.2014.10.001.
- Yanowitz, J., R. L. McCormick, and M. S. Graboski. 2000. In-use emissions from heavy-duty diesel vehicles. Environ. Sci. Technol. 34 (5):729–40. doi:10.1021/es990903w.
- Yu, X.-Y., T. Lee, B. Ayres, S. M. Kreidenweis, J. L. Collett, and W. Malm. 2005. Particulate nitrate measurement using nylon filters. J. Air Waste Manage. Assoc. 55 (8):1100–10. doi:10.1080/10473289.2005.10464721.