
Abstract
Claspin plays multiple important roles in regulation of DNA replication as a mediator for the cellular response to replication stress, an integral replication fork factor that facilitates replication fork progression and a factor that promotes initiation by recruiting Cdc7 kinase. Here, we report a novel role of Claspin in growth recovery from serum starvation, which requires the activation of PI3 kinase (PI3K)-PDK1-Akt-mTOR pathways. In the absence of Claspin, cells do not proceed into S phase and eventually die partially in a ROS- and p53-dependent manner. Claspin directly interacts with PI3K and mTOR, and is required for activation of PI3K-PDK1-mTOR and for that of mTOR downstream factors, p70S6K and 4EBP1, but not for p38 MAPK cascade during the recovery from serum starvation. PDK1 physically interacts with Claspin, notably with CKBD, in a manner dependent on phosphorylation of the latter protein, and is required for interaction of mTOR with Claspin. Thus, Claspin plays a novel role as a key regulator for nutrition-induced proliferation/survival signaling by activating the mTOR pathway. The results also suggest a possibility that Claspin may serve as a common mediator that receives signals from different PI3K-related kinases and transmit them to specific downstream kinases.
INTRODUCTION
Living organisms are exposed to various types of stresses, but they have so-called “stress response systems” to cope with these situations. For example, in the cellular response pathway to replication stress (external and internal conditions where DNA replication is inhibited), a signal is transmitted from sensor kinase (ATR) to effector kinase (Chk1) to temporarily arrest progression of replication and cell division. Claspin functions as a mediator of ATR (a member of PIKK; phosphatidylinositol 3-kinase-related kinase), to Chk1 signaling in the replication stress response.Citation1 The stalled replication fork caused by replication stress results in activation of the ATR-ATRIP complex and activated ATR phosphorylates Chk1 at Ser317 and Ser345 to block DNA replication.Citation2 Chk1 phosphorylation by ATR requires Claspin as a mediator for the checkpoint signal transduction. ATR phosphorylates Chk1 in the Claspin-Chk1 complex; this phosphorylation occurs more efficiently in vitro on the Claspin-Chk1 complex than on the Chk1 alone.Citation3 Although the roles of Claspin and its orthologs in replication checkpoint through interaction with upstream (a member of phosphatidylinositol 3-kinase-related family) and downstream Chk1 kinase effector kinase are well established, its potential roles in other cellular response pathways have not been known. Here, we discovered a novel role of Claspin in the nutrition-induced signaling pathway.
Claspin was originally discovered as a factor that binds to Chk1 and promotes replication checkpoint in Xenopus egg extract.Citation4 Mrc1, its yeast homolog, is also essential for activation of downstream effector kinases (Rad53 and Cds1/Rad53, respectively, in budding and fission yeasts). Thus, the role of Claspin/Mrc1 as a replication checkpoint mediator is well conserved from yeasts to human.Citation3,Citation5–10 The Chk1 binding domain of Claspin (CKBD) binds Chk1 in a manner strictly dependent on its phosphorylation.Citation11 Thr-916 residue in CKBD is phosphorylated after HU (hydroxyurea, an agent that blocks DNA replication by inhibiting ribonucleoside diphosphate reductase) treatment and Chk1 cannot be activated in human cells carrying the T916A/S945A mutation in Claspin.Citation12 It was reported that Cdc7 kinase and casein kinase 1 γ1 are potential kinases for phosphorylating CKBD under DNA replication stress condition.Citation13,Citation14
In addition to the Claspin’s role as a checkpoint adaptor, Claspin may also play important roles in regulation of normal course of DNA replication, similar to Mrc1.Citation8,Citation15–23 Claspin has been shown to be required for efficient DNA replication through its ability to facilitate the replication fork movement.Citation18 We have recently reported a novel function of Claspin in replication initiation control; Claspin promotes phosphorylation of Mcm by recruiting Cdc7, the kinase essential for initiation of DNA replication.
Recent reports show that Mrc1 in budding yeast, the ortholog of Claspin, plays a role in slowing down DNA replication in response to osmotic, oxidative, heat and nutrient deficiency stresses.Citation24,Citation25 In mammalian cells, Claspin is required for suppression of replication origin firing and for slowing DNA replication fork progression in response to endoplasmic reticulum stress.Citation26,Citation27 These cellular pathways are independent of the replication checkpoint pathways, indicating roles of Mrc1/Claspin in stress response pathways other than the replication checkpoint.
Growth factors, like PDGF, FGF and IL3, have been reported to promote cell survival and cell cycle progression.Citation18,Citation28–30 Interactions of these factors with their receptors trigger the activation of phosphoinositide 3-kinase (PI3K).Citation31,Citation32 PI3 kinases are classified into three classes, Class I, Class II and Class III, on the basis of their structure.Citation33 Class I PI3Ks are heterodimers and play important roles in signal transduction. Class I are classified into Class IA and IB. Class IA consists of p110α, β and δ and is associated with the regulatory subunits p85α, p55α, p50α, p85β and p55γ. Class IB PI3K, p110γ, is expressed only in mammals and its function is regulated by the βγ subunit of G proteins and tyrosine kinase receptors.Citation33 Class II consists of α, β, and γ, none of which has regulatory subunits and exhibit enzymatic activity as monomers. The function and activation mechanism of class II are still controversial. PI3K recruits phosphoinositide-dependent protein kinase 1 (PDK1), leading to activation of Akt and mTOR.Citation34–36 Mechanistic target of rapamycin (mTOR), an atypical serine/threonine kinase, belongs to the PIKK. mTORC could accept many intracellular and extracellular signals, such as amino acids, growth factors, energy, oxygen, and DNA damage. Nutrient sensing or responses to nutrient utilization are among of the main functions of the mTORC1, and the signal transmission originating from the levels and quality of nutrients requires the coordinated interaction of sensors and regulators upstream of mTORC1.Citation37 When sufficient nutrients are available, mTOR is phosphorylated at Ser2448 via the PI3K/Akt signaling pathway and transmits signals to downstream factors p70 S6 kinase and eIF4E inhibitor 4EBP1 to promote cell growth and cell cycle progression.Citation38–41 However, potential roles of Claspin in the nutrient-induced PI3K-PDK1-mTOR pathway have not been explored.
In this study, we show that Claspin is required for growth restart from serum starvation-induced growth arrest, and interacts with the ATR-like PIKK, PI3K, and factors in the mTOR pathway in a manner independent of the ATR-Chk1 pathway. Claspin interacts with PI3K, PDK1 and mTOR and promotes phosphorylation of factors involved in the PI3K-PDK1-mTOR pathway, when cells are released from serum starvation. The findings in this study demonstrate a novel role of Claspin in growth recovery from serum starvation as an essential mediator for the PI3K-PDK1-mTOR pathway, and may provide a potential link between DNA replication and nutrition-induced signaling pathway.
RESULTS
Growth recovery from serum starvation requires Claspin
We previously established mouse embryonic fibroblast cells in which Claspin can be inducibly knocked out. In this cell line, the exon 2 are flanked by two loxP sites, and can be excised out by expressing Cre recombinase, leading to knockout of the Claspin gene. We noted that during serum starvation, parts of Claspin knockout MEF cells appeared apoptotic. Upon Claspin knockout, annexin V-positive cells in an early apoptotic stage increased from 5% to 42.9% at 48 h after serum starvation. In TUNEL assays, cells that contain fragmented DNA increased from 10.8% to 64.5% at 48 h after serum starvation. However, analyses of DNA content indicated the sub-G1 population increased only mildly (from 5.3% to 8.1%), indicating that they did not progress into an incorrigible stage ( and ). However, during the growth recovery from the serum starvation, MEF cells lacking the Claspin gene underwent massive cell death ( and ). Furthermore, 293T cells depleted of Claspin by siRNA also similarly died after release from serum starvation ( and ). We examined other replication and checkpoint-related factors for their roles in growth restart after serum starvation. Knockdown of Cdc7 and CKγ1, but not that of Tim, AND1, TopBP1 and Cdc45, caused cell death during serum-induced growth in 293T cells (). This indicates that Claspin’s roles in growth from serum starvation is not simply due to its role in replication checkpoint or to that in replication initiation. We also noted that some population of the surviving Claspin knockout cells became senescent ().
FIG 1 Claspin is required for growth recovery from serum starvation. (A) Claspin (f/+) and (f/-) MEF cells were treated with Ad-Cre or untreated for 48 h. After passage and cell growth for 24 h, cells were incubated in serum-free medium for 0, 24, and 48 h and harvested. Cell apoptosis was analyzed by FACS after annexin V FITC staining. P3, apoptotic cells; P2, nonapoptotic cells. (B) Claspin (f/-) MEF cells were treated with Ad-Cre or untreated for 48 h. After passage and cell growth for 24 h, cells were cultured in serum-free medium for 48 h and harvested. Cell death was analyzed by TUNEL assay. P2, apoptotic cells; P1, nonapoptotic cells. (C) Claspin(+/+), (f/+) and (f/-) MEF cells, treated with Ad-Cre or untreated, were cultured in serum-free medium for 48 h. After serum was added to the medium, the cells were grown for 72 h and observed under microscopy. (D) Quantification of the cell numbers of the cultures in (C). Experiments were repeated three times and error bars are indicated. (E) 293T cells cultured in serum-free medium for 24 h and released into growth by re-addition of serum. Cells were harvested at the indicated times and cell cycle were analyzed by FACS. (F) siControl- or siClaspin-treated 293T cells were cultured in serum-free medium for 24 h and released into growth by re-addition of serum. Cells were observed under microscope at the indicated times. Unsynchronized, cells treated with siControl or siClaspin for 48 h. (G) 293T cells were treated with control siRNA or with siRNA targeting genes indicated for 48 h. Cells were then cultured in serum-free medium for 24 h and released into growth by re-addition of serum. Cell numbers were counted at 24 h after siRNA transfection (–), at 24 h after starvation (starvation), and at 28 h after serum addition (release). Experiments were conducted three times and error bars are indicated. (H) Claspin (f/-) MEF cells treated with Ad-Cre or untreated were cultured in serum-free medium for 48 h, and were released into growth for 0, 26, or 48 h. Photos were taken at each time point, and at 48 h, cells were stained by β-galactosidase and observed under microscope. Left, 4× magnification; right, 20× magnification. Quantification of β-galactosidase positive cells. Experiments were conducted three times and error bars are indicated. (I) Claspin (f/-) MEF cells and the same cells carrying Claspin transgene (WT, DE/A), treated with Ad-Cre or untreated, were cultured in serum-free medium for 48 h. After addition of serum at time = 0, cells, harvested at indicated times, were analyzed by FACS.
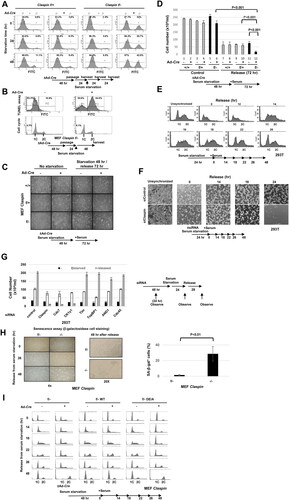
We previously identified an acidic patch segment (AP) on Claspin which is required for replication initiation and replication stress checkpoint functions through its ability to recruit Cdc7 kinase.Citation19 We examined whether AP is required for growth recovery from serum. We ectopically expressed the wild-type and DE/A mutant of Claspin in Claspin (f/-) MEF cells. The DE/A mutant, in which all the acidic residues in AP were replaced with alanine, cannot interact with Cdc7. We analyzed the DNA contents of the cells after release from serum starvation by flow cytometry (). The control cells entered S phase at 14 h after release, and nearly completed it by 22 h, and returned to G1 by 26 h. In contrast, the Claspin knockout cells did not enter S phase or only very slowly replicated its genome by 26 h, and at 48 h, most surviving cells were found to have G2 DNA content. Exogenous expression of the wild-type Claspin restored the timely S phase entry and reversed the cell death effect in the Claspin knockout cells. In contrast, the DE/A mutant could not rescue the defects, indicating that AP is essential for growth restart from serum starvation.
The results described above indicate that Claspin is required for growth and survival from serum starvation, and the acidic patch, known to recruit Cdc7, plays a crucial role for this process.
Effect of Claspin knockout on replication and cell cycle-related factors
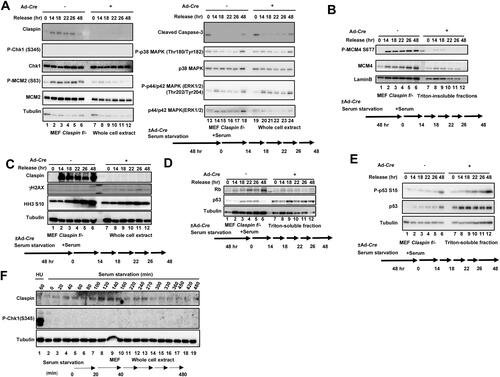
Delayed S phase entry in Claspin knockout may be related to its role in initiation and elongation of DNA synthesis. Therefore, we have examined the factors involved in the process of DNA replication and cell cycle progression during the release from serum starvation. Phosphorylation of MCM (MCM2 (S53) and MCM4 (S6T7)) was induced by 14 h in the control, but was not induced in Claspin knockout, consistent with the fact that cells do not enter S phase in the absence of Claspin ( and ). Histone H3 S10 phosphorylation increased from 18 to 48 h in control, consistent with active cell division, but it did not increase after 22 h in Claspin knockout, consistent with failure of cell division (). Activation of MAP kinases, p38 and ERK1/2, observed during growth stimulation, is not significantly affected by loss of Claspin (). Rb protein increased after growth stimulation in control, but did not increase in Claspin knockout, whereas the total p53 protein as well as the activated form of p53 increased in Claspin knockout, and stayed at the levels higher than that found in the control ( and ). The γH2AX signal moderately increased after release from serum starvation in Claspin knockout cell, indicating the presence of DSB (). However, Chk1 was not activated during serum starvation as well as during release from serum starvation in MEF cells ( and ), indicating that replication checkpoint is not activated. There results indicate that Claspin is required for release from serum starvation through mechanisms that do not involve replication checkpoint activation.
FIG 2 Effects of Claspin knockout on factors involved in DNA replication, cell cycle progression and MAP kinase pathways during the growth recovery from serum starvation. (A to E) Claspin (f/-) MEF cells, treated with Ad-Cre or untreated, were cultured in serum-free medium for 48 h. After addition of serum, cells were harvested at the indicated times and proteins were analyzed by Western blotting with indicated antibodies. (A and C) Whole cell extracts were analyzed. (B, D and E) Cells were fractionated into Triton-soluble and -insoluble fractions, and Triton-insoluble (B) and Triton-soluble fractions (D and E) were analyzed by Western blotting with indicated antibodies. (F) MEF cells cultured in the serum-free medium and harvested at indicated times. Samples were analyzed by Western blotting with the indicated antibodies. The phosphorylated Chk1 (S345) is an indicator for replication checkpoint activation. MEF cells treated with 2 mM HU for 60 min (lane 1) were used as positive control.
Claspin is required for activation of the PI3K/PDK1/mTOR pathway
The PI3K/PDK1/mTOR pathway is intimately related to the nutrient-mediated regulation of cell growth.Citation42 Therefore, we examined the effect of Claspin knockout on this pathway. Expression of PI3 kinase Class III is maintained after release for 26 h and increased during the next 22 h. Conversely, it increased during the first 14 h but decreased after that in Claspin knockout cells. Tyr458 phosphorylation of PI3 kinase p85 subunit was observed at 48 h in control, but was not detected in the Claspin knockout (). S473 phosphorylation of Akt (phosphorylated by mTORC2 and required for its activation) continues to increase after release until 48 h, but that in Claspin knockout slightly increased at 14–18 h and then decreased (). S2448 phosphorylation in mTOR, an indicator of mTOR activation, increased after release and maintained its expression until 48 h after release, but that in Claspin knockout stayed low after release and started to decrease at 18 h (). Phosphorylation of T37/46 of 4EBP1 (a target of mTORC1), which continued to increase in the control, also behaved in a similar manner in Claspin knockout, decreasing at 18 h and thereafter (). However, phosphorylation of S241 of PDK1 (an indicator of active PDK1) increased until 48 h in the wild-type, while it stayed high in the first 22 h and then gradually decreased thereafter in Claspin knockout. In contrast, phosphorylation of S6K T389 was not activated at all in Claspin knockout, whereas it continued to increase until 48 h in control (). These results show that the PI3K/PDK1/mTOR pathway is compromised in Claspin knockout cells during the growth recovery from serum starvation. Thus, Claspin is required for sustained activation of the PI3K/PDK1/mTOR pathway but not for growth-induced MAP kinase pathway during growth stimulation from the quiescent state.
FIG 3 Effects of Claspin knockout on factors involved in the PI3K-Akt-mTOR pathway during the growth recovery from serum starvation. (A) Claspin (f/-) MEF cells, treated with Ad-Cre or untreated, were cultured in serum-free medium for 48 h. Cells were harvested at the indicated times after addition of serum, and the whole cell extracts were analyzed by Western blotting with indicated antibodies. (B) 293T cells were treated with siControl or siClaspin for 24 h, cultured in serum-free medium and harvested at indicated times (left). After 24 h of serum starvation, cells were released by addition of serum and harvested at indicated times (right). The whole cell extracts were analyzed by Western blotting with indicated antibodies. (C) Claspin (f/-) MEF cells exogenously expressing the DE/A mutant were treated with Ad-Cre or untreated and cultured in serum-free medium for 48 h. Growth was restarted by addition of serum and cells were harvested at indicated times. The whole cell extracts were analyzed by Western blotting with indicated antibodies.
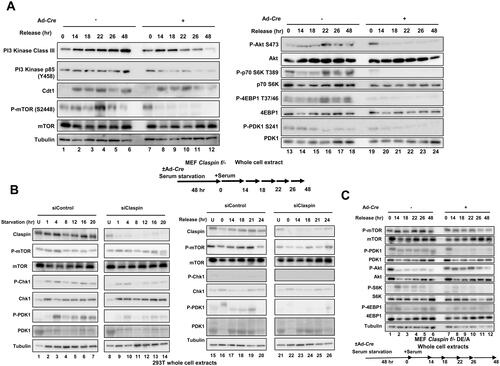
During serum starvation in 293T cells, the levels of phosphorylated PDK1 and mTOR were not affected by Claspin depletion, whereas they decreased in Claspin-depleted cells during the release from serum starvation, as was observed in Claspin knockout MEF cells (). Chk1 was slightly activated during serum starvation, but not activated during the release. Exogenous expression of the DE/A mutant in Claspin knockout MEF cells did not rescue the phosphorylation of PDK1, mTOR, S6K or 4EBP1, indicating that the acidic patch is essential for activation of PI3K/PDK1/mTOR ( and ).
Cell death is induced by loss of Claspin during growth restart from serum starvation and is partially rescued by inhibition of p53 or ROS
Most cells died by 48 h after growth stimulation in the absence of Claspin, as indicated by the FACS analyses ( and ). The p53 level increases and activated during this process (), and indeed, addition of Pifithrin-α, a p53-specific inhibitor, at 50 µM partially rescued the cell death (), indicating that the cell death at least partly depends on p53.
FIG 4 Roles of p53 and ROS in cell death and DNA synthesis during the growth recovery from serum starvation. (A) Claspin (f/-) MEF cells, treated with Ad-Cre, were cultured in serum-free medium for 48 h. After addition of serum and Pifithrin-α, a p53-inhibitor, at various concentrations, cells were harvested at the indicated times and observed under microscope. Viable cell numbers were counted and quantified. Experiments were repeated three times and error bars are indicated. (B) Claspin (f/-) MEF cells, treated with Ad-Cre or untreated, were cultured in serum-free medium for 48 h. After addition of serum, cells were treated with DCFH-DA (2', 7'-dichlorodihydrofluorescin diacetate; ROS indicator) at indicated times and then observed under florescence microscopy. (C) Claspin (f/-) MEF cells, treated with Ad-Cre or untreated, were cultured in serum-free medium for 48 h. Serum was added in the presence or absence of 5 mM NAC, and observed under microscope at 48 h after release. Fractions of live cells were calculated and presented. Experiments were repeated three times and error bars are indicated. (D) Claspin (f/-) MEF cells, treated with Ad-Cre or untreated, were cultured in serum-free medium for 48 h. At t = 0, serum was added with or without NAC, and BrdU was added to the cells for 30 min at the indicated times. DNA contents were analyzed by FACS. (E) Quantification of BrdU incorporation from the data in (D). Experiments were conducted three times and error bars are indicated.
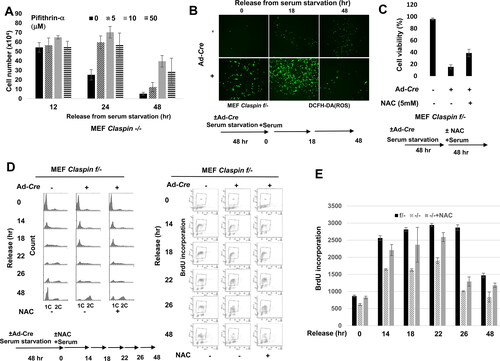
ROS (reactive oxygen species) often becomes a primary cause for genomic instability. The mTORC1 repressors such as Sesn1, TSC2, AMPKβ1, and IGF-BP3 also play roles in the regulation of ROS.Citation43,Citation44 p53 activates these mTORC1 repressors, resulting in lower metabolic activity and increased oxidative stress.Citation45 Therefore, we examined production of ROS after release from serum starvation. In Claspin knockout MEF cells, more ROS were produced after serum starvation, and the level of ROS further increased at 18 h after release (). We then examined the effect of NAC, anti-ROS reagent, for its ability to suppress cell death. At 5 mM, NAC restored the cell viability up to 40% compared to less than 20% in nontreated cells (). It also partially restored cell cycle progression and DNA synthesis ( and ). These results indicate ROS, generated in Claspin knockout cells, are also at least partially responsible for cell death.
Claspin interacts with PI3 kinase, mTOR, and PDK1
In order to understand how Claspin may function in the PI3K/PDK1/mTOR pathway, we examined whether Claspin physically interacts with the factors involved in this pathway. We expressed Flag-tagged Claspin transiently in 293T cells, and immunoprecipitated with antibody against the Flag tag. HA-tagged Claspin was used as a negative control. PI3 kinase p100β as well as mTOR were efficiently pulled down, and PI3 kinase Class III and PI3 kinase p100α were also coimmunoprecipitated (), although the efficiency was lower. The phosphorylated form of PDK1 (S241) also interacted with Claspin. In contrast, Akt, S6 kinase and 4EBP1 did not interact with Claspin (). Furthermore, we show that endogenous Claspin also interacts with PI3 kinase p100β, PI3 kinase Class III, PDK1 S241 and mTOR ().
FIG 5 Claspin interacts with members of the PI3K-PDK1-mTOR pathway. (A) Flag-tagged wild-type or HA-tagged (negative control) Claspin was expressed in the 293T cells, and immunoprecipitates pulled down by anti-Flag antibody agarose beads were analyzed by Western blotting with indicated antibodies. (B) Triton-soluble extracts of 293T were prepared and were used for immunoprecipitation by anti-IgG or anti-Claspin antibody. The input extracts and immunoprecipitates were analyzed by Western blotting with antibodies indicated. (C) The schematic drawing of the wild-type and mutant Claspin polypeptides used in the pull-down assays. (D) Flag-tagged wild-type or various mutant Claspin polypeptides were expressed in 293T cells, and immunoprecipitates pulled down by anti-Flag antibody agarose beads were analyzed as in (A). Among PI3 kinase subtypes, PI3 kinase p100β, that showed strongest binding, was examined. (E) 293T cells cultured in serum-free medium for 24 h and released by addition of serum were harvested at indicated times. Triton-soluble extracts were prepared and were used for immunoprecipitation by anti-Claspin antibody. The input extracts and immunoprecipitates were analyzed by Western blotting with antibodies indicated. UN, untreated. Lane 8, immunoprecipitated by control IgG. (F) 293T cells were cultured in normal medium (control) or serum-free medium for 24 h (serum starvation 24 h). Cells in serum-free medium were released by addition of serum for 14 h (Release 14 h). Cells were fixed with PFA, treated with Triton X100, and stained with anti-mouse Alexa 488 conjugated anti-Claspin antibody (green) and anti-rabbit Alexa 594 conjugated anti-P-mTOR antibody (phosphorylated at S2448; red). Cells were observed under fluorescence microscopy. (G) Quantification of the fractions of P-mTOR signals overlapping with Claspin signals in cells under normal growth, after starvation, or after release from starvation, based on the data in (F). We counted the number of P-mTOR-positive cells expressing P-mTOR in cytosol and/or nuclei excluding dead cells. We then counted cells with nuclei of yellow signals (overlapping of P-mTOR [Red] and Claspin [Green]) and divided by the numbers of the P-mTOR-positive cells. We counted ∼300 cells in each experiment. Experiments were repeated three times and error bars are indicated. (H) HCT116 and U2OS cells were cultured in normal medium (C) or serum-free medium for 24 h (S), and released for 14 h (R). Claspin and phosphorylated mTOR were detected as described in (F), and the fractions of P-mTOR signals that overlap with the Claspin signals were quantified as described in (G). Experiments were conducted three times and error bars are indicated. (I) Various Flag-tagged Claspin polypeptides were mixed with purified PDK1 in vitro, and pulled down by anti-Flag agarose beads, followed by analysis by Western blotting with indicated antibodies. (J) Purified PDK1 was incubated with various purified Flag-tagged Claspin polypeptides, pulled down by anti-Flag agarose beads and was analyzed by Western blotting with indicated antibodies. One fifth of the reaction mix before pull-down is analyzed as Input. In ST27/A, ST5/A and ST19/A, all the serines/threonines in aa903-1120, aa1121-1218, and aa1219-1337, respectively, were replaced by alanine. ST27/E has glutamic acids replacing all the serines/threonines in aa903-1120, while all the acidic residues (aspartic acid and glutamic acid) were replaced with alanine in 988-1086 aa in DE/A.
![FIG 5 Claspin interacts with members of the PI3K-PDK1-mTOR pathway. (A) Flag-tagged wild-type or HA-tagged (negative control) Claspin was expressed in the 293T cells, and immunoprecipitates pulled down by anti-Flag antibody agarose beads were analyzed by Western blotting with indicated antibodies. (B) Triton-soluble extracts of 293T were prepared and were used for immunoprecipitation by anti-IgG or anti-Claspin antibody. The input extracts and immunoprecipitates were analyzed by Western blotting with antibodies indicated. (C) The schematic drawing of the wild-type and mutant Claspin polypeptides used in the pull-down assays. (D) Flag-tagged wild-type or various mutant Claspin polypeptides were expressed in 293T cells, and immunoprecipitates pulled down by anti-Flag antibody agarose beads were analyzed as in (A). Among PI3 kinase subtypes, PI3 kinase p100β, that showed strongest binding, was examined. (E) 293T cells cultured in serum-free medium for 24 h and released by addition of serum were harvested at indicated times. Triton-soluble extracts were prepared and were used for immunoprecipitation by anti-Claspin antibody. The input extracts and immunoprecipitates were analyzed by Western blotting with antibodies indicated. UN, untreated. Lane 8, immunoprecipitated by control IgG. (F) 293T cells were cultured in normal medium (control) or serum-free medium for 24 h (serum starvation 24 h). Cells in serum-free medium were released by addition of serum for 14 h (Release 14 h). Cells were fixed with PFA, treated with Triton X100, and stained with anti-mouse Alexa 488 conjugated anti-Claspin antibody (green) and anti-rabbit Alexa 594 conjugated anti-P-mTOR antibody (phosphorylated at S2448; red). Cells were observed under fluorescence microscopy. (G) Quantification of the fractions of P-mTOR signals overlapping with Claspin signals in cells under normal growth, after starvation, or after release from starvation, based on the data in (F). We counted the number of P-mTOR-positive cells expressing P-mTOR in cytosol and/or nuclei excluding dead cells. We then counted cells with nuclei of yellow signals (overlapping of P-mTOR [Red] and Claspin [Green]) and divided by the numbers of the P-mTOR-positive cells. We counted ∼300 cells in each experiment. Experiments were repeated three times and error bars are indicated. (H) HCT116 and U2OS cells were cultured in normal medium (C) or serum-free medium for 24 h (S), and released for 14 h (R). Claspin and phosphorylated mTOR were detected as described in (F), and the fractions of P-mTOR signals that overlap with the Claspin signals were quantified as described in (G). Experiments were conducted three times and error bars are indicated. (I) Various Flag-tagged Claspin polypeptides were mixed with purified PDK1 in vitro, and pulled down by anti-Flag agarose beads, followed by analysis by Western blotting with indicated antibodies. (J) Purified PDK1 was incubated with various purified Flag-tagged Claspin polypeptides, pulled down by anti-Flag agarose beads and was analyzed by Western blotting with indicated antibodies. One fifth of the reaction mix before pull-down is analyzed as Input. In ST27/A, ST5/A and ST19/A, all the serines/threonines in aa903-1120, aa1121-1218, and aa1219-1337, respectively, were replaced by alanine. ST27/E has glutamic acids replacing all the serines/threonines in aa903-1120, while all the acidic residues (aspartic acid and glutamic acid) were replaced with alanine in 988-1086 aa in DE/A.](/cms/asset/3797edc7-e59f-43b6-ac78-ba7267e4152d/tmcb_a_2160598_f0005_c.jpg)
Next we examined which domain(s) of Claspin is (are) required for interaction with these factors involved in the nutrition-induced signaling pathway. The segment containing acidic patch interacts not only with Cdc7 but also with a region of Claspin in the N-terminal region, inhibiting DNA and PCNA binding activities of Claspin.Citation18 Various truncated segments of Claspin, C-terminally Flag-tagged, were expressed in 293T cells and immunoprecipitates were prepared by Flag antibody (). The 897-1101 aa polypeptide (#27) of Claspin could bind to mTOR and PDK1 and the 897-1209 aa polypeptide (#13) could bind to PI3 kinase p110β (, lanes 21 and 20). The N-terminal polypeptides of Claspin (#2 and #26) could not bind to PI3 kinase p100β, mTOR or PDK1 (, lanes 24 and 18). Interactions of PI3 kinase p110β, mTOR and PDK1 with #18 (897-1339 aa) and #24 (1-350 aa + 897-1339 aa) were weaker than that with #13 (897–1209 aa) and #19 (1–350 aa + 897–1209 aa), respectively (, lanes 19/20 and 23/22). This suggests a possibility that the C-terminal segment (1210-1339) may be inhibitory for the interaction of Claspin with PI3 kinase p110β, mTOR and PDK. However, we cannot rule out the possibility that the inhibition is due to the change of the overall protein structures caused by the deletion. mTOR interacts with ST27/E strongly (lane 17), suggesting that prior phosphorylation of Claspin may play a role in its interaction with mTOR.
During the recovery from serum starvation in 293T cells, Claspin interacted with mTOR. The interaction peaked at 14 h after the release and decreased afterward. However, interaction of Claspin with PI3K was constitutively observed during the recovery from the serum starvation (). These results suggest a possibility that Claspin contributes to activation of mTOR pathway through direct protein–protein interactions.
Immunostaining indicates that mTOR is present generally in cytoplasm in asynchronously growing cells as well as in serum-starved 293T cells, whereas Claspin is present in nuclei. Upon release from the serum starvation for 14 h, activated mTOR relocated to nuclei, overlapping with Claspin ( and ). Similar overlap of Claspin and activated mTOR was also observed in U2OS and HCT116 cells released from serum starvation (). Nuclear localization of mTOR was previously reported in many cell lines including normal human fibroblasts.Citation46
PDK1 binds to phosphorylated CKBD of Claspin and facilitates binding of mTOR to Claspin
We also investigated if Claspin directly binds to PDK1 by pull-down experiments using purified Claspin and PDK1 proteins. The results indicate that Claspin directly interacts with PDK1 (, lane 12 and , lane 18). A C-terminal polypeptide of Claspin (#13) bound to PDK1 directly, but N-terminal polypeptides of Claspin (#2 or #26) did so only very weakly (, lanes 16, 18 and 20). The mutant Claspin harboring ST27/A or ST27/E mutations in the C-terminal segment bound to PDK1 as efficiently as the wild-type except for ST27/A which showed reduced binding to PDK1 (, lanes 22, 24, 26 and 28). In contrast, the acidic domain mutant, DE/A, bound to PDK1 only weakly (, lane 20), largely consistent with the in vivo interaction (, lane 15). Furthermore, there is a PDK1 binding site (FXXF;Citation47) in the aa 998-1001 of the acidic domain (FGDF). These results suggest that PDK1 directly recognizes and binds to the acidic patch segment. Alternatively, PDK1 may bind preferentially to the phosphorylated forms of Claspin, since the DE/A mutant fails to recruit kinases such as Cdc7 or CK1, and is expected to be a hypo-phosphorylated form (see below).
Phosphorylated CKBD of Claspin recruits Chk1 kinase in replication checkpoint. Chk1 and PDK1 share 27.5% similarity (47.5% if confined to the N-terminal 210 amino acids) in their kinase domains. Therefore, we decided to examine the possibility that PDK1 is recruited to CKBD in a phosphorylation-dependent manner. The phosphorylated CKBD polypeptide containing two critical phosphorylation sites (T916 and S945) pulled down purified PDK1 (, lane 8). Shorter polypeptides containing only pT916 or pS945 also pulled down PDK1, albeit with reduced efficiency (, lanes 10 and 12). With all the three peptides, prior dephosphorylation of the peptide eliminated the binding (, lanes 9, 11 and 13), indicating that PDK1 binds to CKBD of Claspin in a phosphorylation-dependent manner. This shows the importance of CKBD phosphorylation in recruitment of PDK1 to Claspin, and suggests a possibility that PDK1 acts as an effector kinase for Claspin-mediated mTOR activation during serum-induced growth restart.
FIG 6 Physical and functional interactions between Claspin, PDK1, PI3K and mTOR. (A) Biotin-tagged CKBD peptides as indicated, treated with λPPase or not, were mixed with purified PDK1, and were pulled down by streptavidin conjugated beads. The PDK1 co-pulled down with CKBD was analyzed by Western blotting. (B) Purified His-tagged 897-1209 aa Claspin polypeptides (wild-type, ST27/A and ST27/E mutant) were mixed with PDK1, mTOR (upper panel) or PIK3CB, PIK3C3 (lower panel) proteins and kinase assays were conducted. Samples were analyzed on SDS-PAGE, dried and the gel was autoradiographed (left panels). Middle panels, CBB staining of the same gels. Right drawing, schematic representation of Claspin mutants of 897-1209 polypeptides. The arrowhead indicates the position of 897-1209 polypeptides. The signals above the Claspin bands in lanes 5–8 represent auto-phosphorylated PDK1 which migrates differentially on SDS-PAGE depending on the extent of phosphorylation. (C) The 897-1209 aa Claspin polypeptides (wild-type and ST27/A mutant) were incubated with purified PDK1 in the presence of PI3KB, Cdc7 or CK1γ1 under the kinase reaction condition and were pulled down by Ni-NTA agarose. The co-pulled down PDK1 was analyzed by Western blotting (lower panel). Upper panel, input. (D) Purified mTOR-Flag was incubated with full-length Flag-tagged Claspin polypeptides (WT [full-length] and #2 [1–350 aa]), pulled down by anti-Claspin (N-terminal) antibody, and the IP was analyzed by Western blotting with anti-Flag antibody. One fifth of the reaction mix before pull-down was analyzed as Input. (E) Biotin-tagged CKBD peptides, treated with λPPase or not, were mixed with purified mTOR protein, and were pulled down by streptavidin conjugated beads. The mTOR co-pulled down with CKBD was analyzed by Western blotting. (F) Flag-tagged Claspin was overexpressed in 293T, and was treated with indicated inhibitors. The proteins pulled down by anti-Flag antibody beads were analyzed by Western blotting with the antibodies indicated.
![FIG 6 Physical and functional interactions between Claspin, PDK1, PI3K and mTOR. (A) Biotin-tagged CKBD peptides as indicated, treated with λPPase or not, were mixed with purified PDK1, and were pulled down by streptavidin conjugated beads. The PDK1 co-pulled down with CKBD was analyzed by Western blotting. (B) Purified His-tagged 897-1209 aa Claspin polypeptides (wild-type, ST27/A and ST27/E mutant) were mixed with PDK1, mTOR (upper panel) or PIK3CB, PIK3C3 (lower panel) proteins and kinase assays were conducted. Samples were analyzed on SDS-PAGE, dried and the gel was autoradiographed (left panels). Middle panels, CBB staining of the same gels. Right drawing, schematic representation of Claspin mutants of 897-1209 polypeptides. The arrowhead indicates the position of 897-1209 polypeptides. The signals above the Claspin bands in lanes 5–8 represent auto-phosphorylated PDK1 which migrates differentially on SDS-PAGE depending on the extent of phosphorylation. (C) The 897-1209 aa Claspin polypeptides (wild-type and ST27/A mutant) were incubated with purified PDK1 in the presence of PI3KB, Cdc7 or CK1γ1 under the kinase reaction condition and were pulled down by Ni-NTA agarose. The co-pulled down PDK1 was analyzed by Western blotting (lower panel). Upper panel, input. (D) Purified mTOR-Flag was incubated with full-length Flag-tagged Claspin polypeptides (WT [full-length] and #2 [1–350 aa]), pulled down by anti-Claspin (N-terminal) antibody, and the IP was analyzed by Western blotting with anti-Flag antibody. One fifth of the reaction mix before pull-down was analyzed as Input. (E) Biotin-tagged CKBD peptides, treated with λPPase or not, were mixed with purified mTOR protein, and were pulled down by streptavidin conjugated beads. The mTOR co-pulled down with CKBD was analyzed by Western blotting. (F) Flag-tagged Claspin was overexpressed in 293T, and was treated with indicated inhibitors. The proteins pulled down by anti-Flag antibody beads were analyzed by Western blotting with the antibodies indicated.](/cms/asset/4fa48e69-f196-4885-8295-a9caab17036d/tmcb_a_2160598_f0006_c.jpg)
What would be the kinase responsible for phosphorylation of CKBD? We have examined the possibility that mTOR or PI3K may act as an initial kinase that stimulates PDK1 activation. Purified phosphoinositide-3-kinase beta (PIK3CB: p110β) or phosphoinositide-3-kinase, class III (PIK3C3) phosphorylated the Claspin polypeptide (897–1209 aa) in vitro. The phosphorylation was significantly reduced by substitution of serine/threonine with alanine in the 903-1120 aa segment of Claspin (, lanes 18, 19, 22 and 23). Furthermore, PDK1, like PIK3CB and PIK3C3, also phosphorylated the 897–1209 aa segment of Claspin (, lanes 6–8). To investigate whether Claspin phosphorylated by PIK3CB promotes recruitment of PDK1 or not, we mixed his-tagged Claspin polypeptide (897-1209 aa) and PDK1 in the presence of PIK3C3 or Cdc7 or CK1, the latter two of which were reported to phosphorylate CKBD of Claspin.Citation13 The Claspin polypeptide pulled down by Ni-NTA column was examined for the presence of PDK1. PDK1 interacted with the Claspin polypeptide with efficiency ∼3 times better with PIK3C3 than with Cdc7 or CK1 or without any kinase (, lanes 17, 21, 24, and 27). Furthermore, the same polypeptide of Claspin in which serine/threonine residues in 903-1120 were replaced with alanine, interacted with PDK1 very weakly (, lanes 18, 19, 22, 25 and 28). These results indicate that PIK3B-mediated phosphorylation of the 903-1120 aa segment of Claspin promotes recruitment of PDK1 to Claspin. In contrast, purified mTOR phosphorylated Claspin 897-1209 aa polypeptide in a manner independent of serine/threonine residues in the 903-1120 aa segment (, lanes 10–12), suggesting that the 11211209 aa segment of Claspin may be phosphorylated by mTOR.
Purified mTOR bound to the full-length Claspin, but not to the N-terminal polypeptide of Claspin (#2) (, lane 10), suggesting that Claspin can directly interact with mTOR through its C-terminal segment. Indeed, mTOR was pulled down by the CKBD polypeptides, and this interaction was only partially affected by phosphorylation state of the CS/TGK, the target of PDK binding (). It is likely that mTOR binds to the C-terminal segments of Claspin including CKBD following the recruitment of PDK1 to the phosphorylated CKBD. PDK1-mediated phosphorylation of the C-terminal segment may stimulate the binding of mTOR to Claspin. This prediction is supported by the fact that the ST27/E mutant of Claspin can pull down mTOR most efficiently (, lane 17). Furthermore, binding of mTOR to Claspin was inhibited by the presence of GSK2334470, a PDK1 inhibitor (, lane 9), supporting the role of PDK1 in recruitment of mTOR to Claspin.
These results support a model that PDK1 binds to CKBD of Claspin which may be phosphorylated by PI3K and this facilitates binding of mTOR to Claspin.
Modification of Claspin and other factors during survival under serum starvation and following growth recovery
In both MEF and 293T cells, Claspin is mobility-shifted on SDS-PAGE after serum starvation for 24 h and the shift disappeared after release by addition of serum (, lane 1 and , lane 3). Treatment with λPPase eliminated the mobility-shift (, lane 2), indicating that the shift is caused by phosphorylation. Through mass-spec analyses, we have identified clusters of amino acids in the N-terminal and C-terminal segments of Claspin that are phosphorylated in response to serum starvation ().
FIG 7 Serum starvation induces Claspin phosphorylation by PI3K pathway. (A) 293T cells were starved for serum for 24 h. The whole cell extracts were analyzed by Western blotting using anti-Claspin antibody. (B) 293T cells were serum-starved for 24 h, released for 14 h and harvested. Triton-soluble extracts were prepared from serum-starved (24 h) cells and from starved-and-released (14 h) cells. They were treated with λPPase for 30 min (+) or untreated (–), and then analyzed by western blotting with anti-Claspin antibody. (C) Phosphorylation sites of Claspin in the nontreated cells and in serum-starved (24 h) cells were determined by mass spectrometry analyses and are shown on the drawing along the polypeptide map. Phosphorylation sites identified are indicated by small bars and numbers. Red and blue numbers indicate those unique to each condition and those common to both conditions, respectively. Three Chk1 binding domains (CKBD) and AP (acidic patch) are shown by yellow boxes and by a purple box, respectively. (D and E) 293T cells were starved for serum for 24 h, and inhibitors shown were added during the last 12 h before the harvest. The whole cell extracts were analyzed by Western blotting with the antibodies indicated. DMSO was added as a control. (F) 293T cells were treated with siControl or siClaspin for 24 h in normal medium (Control), or in serum-free medium (Starvation). Alternatively, cells, after 24 h serum starvation, were released for 12 h (Release 12 h) or 24 h (Release 24 h). Inhibitors, as indicated, or DMSO (control) were added for the final 12 h before the harvest. Cells were observed under microscope and live cell numbers were counted. Experiments were conducted three times and error bars are indicated.
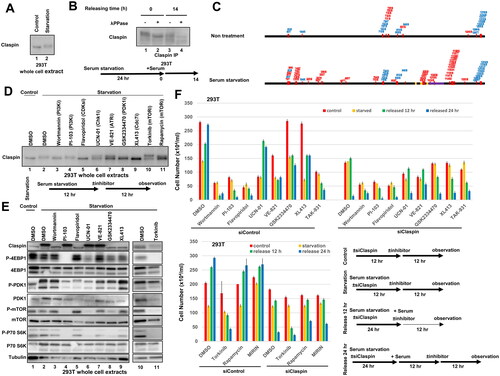
Experiments with various kinase inhibitors indicated that Wortmannin (PI3K, DNA-PK/ATM inhibitor), Flavopiridol (CDKs inhibitor), GSK2334470 (PDK1 inhibitor) and XL413 (Cdc7 inhibitor) reduced the level of mobility-shift of Claspin (, lanes 3, 5, 8 and 9). CDK has been reported to activate mTOR to promote cell growth.Citation48 Inhibition of CDK activity by flavopiridol did not affect the mTOR activity but reduced PDK1 activity at the time of starvation (, lane 5), and induced strong cell death (). Wortmannin (PI3K, DNA-PK/ATM inhibitors) as well as Torkinib (mTOR inhibitor) caused massive cell death during growth recovery. Torkinib reduced mTOR activation and S6K/4EBP1 phosphorylation at the time of serum starvation (, lane 11).
GSK2334470 largely eliminated starvation-induced Claspin phosphorylation (, lane 8), but did not induce cell death (), and only partially inhibited mTOR and downstream kinases at the time of starvation (, lane 8). Cdc7 promotes DNA replication and cope with stalled forks by phosphorylating Mcm and Claspin, respectively.Citation13,Citation18 Cdc7 inhibitor, XL413, also inhibited Claspin phosphorylation ( and , lane 9) but did not affect the PDK1-mTOR activity under serum starvation stress (, lane 9), whereas it induced cell death after serum stimulation (). In contrast, PI3K inhibitor PI-103 did not affect Claspin phosphorylation under serum starvation but induced cell death after release from serum starvation ( lane 4; ), and suppressed activities of mTOR and downstream kinases at the time of starvation (, lane 4).
The results of experiments using inhibitors show that Claspin is phosphorylated at specific residues during serum starvation through actions of several kinases including Cdk, Cdc7 and PIKK, and support the important roles of the PI3K-mTOR pathway in serum induced growth and survival from serum starvation.
PDK1 is mobility-shifted on SDS-PAGE during serum starvation (, lane 2). λPPase did not affect the mobility shift (data not shown), suggesting that PDK1 undergoes some other mode of post-translational modification under serum starvation, the nature of which is currently unknown.
CKBD of Claspin is required for survival and the activation of the mTOR pathway after serum starvation
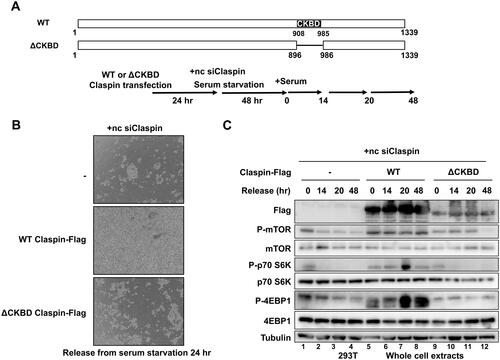
Our results indicate that Claspin plays an important role in nutrition-induced signaling by activating the mTOR pathway to increase cell survival, and a potential important role of CKBD segment in this pathway. In order to evaluate the roles of CKBD in serum-induced growth and survival, we expressed Flag-tagged wild-type or CKBD deletion (ΔCKBD) Claspin in 293T cells and investigated the cell survival and mTOR activation after release from serum starvation (). Upon Claspin knockdown, massive cell death occurred in control cells (, upper panel). This cell death was rescued by expression of the wild-type Claspin (, middle panel). Alternatively, ΔCKBD Claspin did not fully protect Claspin KD cells from cell death (, lower panel). Furthermore, mTOR-S6K-4EBP1 pathway, which was completely inactivated by Claspin knockdown, was reactivated by expression of the wild-type Claspin, but not by that of ΔCKBD (). These results demonstrate that CKBD of Claspin is required for survival and activation of mTOR and its downstream targets.
FIG 8 CKBD of Claspin is required for cell survival and the activation of the mTOR pathway during the release from serum starvation. (A) The schematic drawing of the wild-type and ΔCKBD (Δ895–985 aa) Claspin proteins. (B) 293T cells were transfected with no DNA, Flag-tagged wild-type (WT), or ΔCKBD Claspin for 24 h, and then were transfected with noncoding Claspin siRNA (nc siClaspin) for 48 h concomitant with serum deprivation. Serum was added to the medium at time = 0, and the cells were grown for 24 h, followed by observation under microscopy. (C) Cells from (B) were harvested at 0, 14, 20, 48 h after addition of serum and the whole cell extracts were analyzed by Western blotting with indicated antibodies.
DISCUSSION
Cellular response to metabolic and environmental changes is one of the most important biological processes in cell proliferation and survival. Although detailed studies have been conducted on cellular responses induced by various types of external stress or stimuli, how these cellular responses cross talk and control cell proliferation and survival in an integrated manner has not been fully elucidated.
Claspin/Mrc1 is a conserved protein that mediates replication stress signal to a downstream effector kinase. Budding yeast Mrc1 has been implicated in cellular responses to osmotic, oxidative, heat and nutrient stresses in a manner independent of the replication checkpoint pathway.Citation25 However, roles of Claspin in responses to similar biological stresses/stimuli have been elusive in higher eukaryotes. Here, we found that Claspin plays a novel and important role in cellular responses during the growth recovery from serum starvation.
Claspin is required for DNA replication and survival during growth restart from serum starvation: roles in activation of PI3K-PDK1-mTOR
In Claspin knockout MEF cells, cells show reduced DNA synthesis, consistent with its role in facilitating the phosphorylation of Mcm by Cdc7 kinase. In release from serum starvation, Claspin is required for DNA synthesis, presumably reflecting its role in initiation of DNA replication. More strikingly, Claspin is required for activation of the PI3K-PDK1-mTOR pathway. The loss of the PI3K activation is not due to impaired initiation, since depletion of Cdc45, known to be essential for initiation, did not affect the viability of the cells (). Rather, Claspin is likely to be involved in activation of the PI3K-PDK1-mTOR pathway through direct interaction with these proteins. The DE/A mutant interacted with PDK1, mTOR or PI3K only with reduced efficiency, and did not rescue the cell death in Claspin knockout cells ( and ). We also showed that CKBD is required for cell survival and mTOR activation during growth restart from serum starvation (). These proteins exhibited similar binding pattern to the panels of deletion derivatives of Claspin. They interact with Claspin through aa897–1209 carrying CKBD + AP segment ( and).
We discovered that PDK1 binds to CKBD in a manner dependent on the phosphorylation of the critical residue in the CKBD. This is reminiscent to binding of Chk1 to the phosphorylated CKBD in response to replication stress. PDK1 may bind to phosphorylated CKBD of Claspin during recovery from serum starvation. This binding is required for further activation of mTOR, since a PDK inhibitor, GSK2334470, reduced the interaction between Claspin and mTOR (, lane 9). Interaction of mTOR with Claspin was stimulated by the substitution of serine/threonines in the C-terminal segment by glutamic acids (; ST27/E, lane 17). This suggests that phosphorylation of this segment of Claspin, presumably by PDK1 recruited to CKBD, facilitates the recruitment of mTOR to Claspin. In contrast, PI3K may play a role in binding of PDK1 to Claspin, since PI3K promotes the binding of PDK1 to Claspin in vitro (). However, Cdc7 or CK1γ1, which phosphorylates CKBD of Claspin, did not promote PDK1 binding to Claspin. This suggests that phosphorylation of residues other than CKBD in the 897-1209 aa segment also contributes to interaction of Claspin with PDK1.
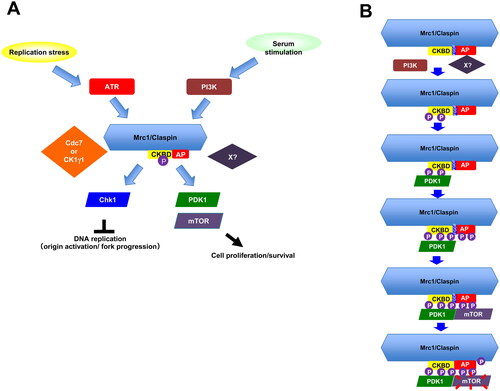
On the basis of these data, we propose that during the recovery from serum starvation, PI3K interacts with Claspin and phosphorylates the critical residues on CKBD and other segment of the 899-1120 aa region. This would recruit PDK1, which then phosphorylates Claspin, facilitating the interaction of mTOR with Claspin. mTOR interacts with non-phosphorylated form of CKBD, suggesting that the requirement of phosphorylation for mTOR interaction may not be absolute. These results suggest a possibility that Claspin may act as a platform for the activation of the PI3K-PDK1-mTOR pathway (). Currently, we do not know exactly the nature of the kinases responsible for phosphorylation of Claspin required for interaction with PDK1. Although PI3K is a prime candidate, we cannot rule out the possibility of involvement of other kinases. Cdc7 knockdown or inhibition by inhibitors induced cell death after release from starvation in 293T cells ( and ). This suggests a possibility that Cdc7 kinase (and CK1γ1) also contribute to phosphorylation of Claspin during growth restart.
FIG 9 Mrc1/Claspin in serum-stimulated growth: a model (A) In response to serum stimulation, PI3K is activated and phosphorylates Claspin in its C-terminal region. This may facilitate the binding of PDK1 to Claspin and in turn promote binding of mTOR to Claspin, leading to activation of mTOR. Thus, PI3K-Claspin-PDK1-mTOR pathway functions for cell growth and survival after serum stimulation. The proposed pathway is highly reminiscent to the ATR-Claspin-Chk1 pathway activated by replication stress. (B) Serum stimulation activates PI3K, which would phosphorylate Claspin. Another unknown factor X, that would be directly recruited by AP, may also be involved in this step. Claspin, phosphorylated in its C-terminal segment including CKBD, now recruits PDK1 in a manner dependent on the phosphorylation of CKBD. This step may be facilitated also by the FGDF sequence (aa 998-1001), a putative binding sequence of PDK1. PDK1 phosphorylates Claspin further in its C-terminal segment and this would in turn recruits mTOR on Claspin. mTOR is now activated, induces downstream pathway and phosphorylates the segment further C-terminal to AP.
Potential activation of PI3K-PDK1-mTOR in nuclei
Activation of the PI3K pathway is a cytoplasmic event. Claspin is a nuclear protein and indeed it is detected in nuclei in 293T cells (). How does nuclear Claspin interact with the factors involved in the PI3K-PDK1-mTOR pathway? It was reported that PI3Kβ is present also in nuclei and the nuclear PI3Kβ is essential for cell viability.Citation49 PI3Kβ was reported to interact with PCNA and to be required for chromatin binding of PCNA.Citation50 mTOR was reported to be predominantly localized in nuclei in many cell lines examined including normal human fibroblasts.Citation46 Thus, Claspin would activate the PI3K-PDK1-mTOR pathway in nuclei. This could contribute to the process of DNA replication and/or to the completion of mitosis.
Loss of Claspin during growth restart from serum starvation leads to ROS and p53-dependent cell death
The transcription factor p53 is a major regulator of the DNA damage response and mTOR signaling.Citation51–53 The role of p53 as a responder to DNA damage may be pre-death or postsurvival, depending on the extent, timing and cell type of DNA damage. Typically, severe DNA damage results in sustained activation of p53, while minor DNA damage activates p53 only briefly. Thus, the former results in cell death, while the latter induces repair and survival mechanisms.Citation54 p53 is an mTOR antagonist, and thus, in response to cellular stress, p53 inhibits cell proliferation and suppresses mTOR to trigger DNA damage response. Conversely, in a negative feedback loop, mTOR increases p53 activity.Citation55 Furthermore, p53 also functions to deal with ROS.Citation56 The link between ROS and p53 is complex. ROS levels are generally high in cancer cells compared to normal cells due to their hypermetabolism, and the high ROS levels may be responsible for instability in the genome that leads to tumor formation.Citation57 Claspin depletion leads to failure to activate the PDK1-mTOR pathway, eventually leading to impaired DNA replication or unattended replication-transcription collisions. This would generate a large amount of ROS and activate p53, leading to cell death.
Potential mechanism of Claspin-mediated activation of the mTOR pathway in response to nutritional stress
mTOR coordinates eukaryotic cell growth and metabolism in response to microenvironmental stimuli, including nutrients and growth factors. Extensive research during the past decades has shown that mTOR plays a crucial role also in the regulation of fundamental cellular processes such as protein synthesis, autophagy, and oxidative stress responses and that deregulated mTOR signaling is related to cancer progression and aging.Citation58
mTOR regulates many of the components involved in initiation and elongation stages of protein synthesis, as well as in the biogenesis of the ribosome itself.Citation59,Citation60 mTOR also mediates translational controlCitation61 and metabolism.Citation58 Gene ontology analysis (GO) indicated that genes involved in translation, peptide metabolism and peptide biosynthesis are among the specifically suppressed in Claspin knockout MEF cells (data not shown). These results support the conclusion that Claspin plays crucial roles in cell growth and survival specifically during serum starvation and during growth restart from serum starvation through activation of the PI3K-mTOR pathway.
The Claspin-PI3K/PDK1/mTOR interaction is crucial for maintaining the growth from serum starvation. How does Claspin facilitate the PI3K-PDK1-mTOR pathway during growth from serum starvation? We envision two possibilities. One is that Claspin functions as an essential mediator that transmits the upstream PIKK signal to downstream effector kinases through phosphorylation relay. The other is that Claspin serves as a molecular scaffold on which the PI3K-PDK1-mTOR factors assemble and are efficiently activated. The role of such a scaffold protein has been proposed also for other signal transduction pathways or cellular reactions.Citation62,Citation63
MATERIALS AND METHODS
Cell lines
293T, U2OS and HCT116 were obtained from ATCC. Mouse embryonic fibroblasts (MEFs) were established from E12.5 embryos as described.Citation19 HCT116 were cultured at 37 °C in a 5% CO2 humidified incubator in McCoy's 5A (modified) Medium (Thermo Fisher) supplemented with 10% fetal bovine serum (Gibco) and 100 U/mL penicillin and 100 μg/mL streptomycin. 293T, U2OS and MEF cells were cultured at 37 °C in a 5% CO2 humidified incubator in Dulbecco’s modified Eagle’s medium (high glucose) (Nacalai Tesque Inc.) supplemented with 15% fetal bovine serum (HANA-NESCO), 2 mM L-glutamine (Gibco), 1% sodium pyruvate (Nacalai Tesque Inc.), 100 U/mL penicillin and 100 μg/mL streptomycin.
Plasmid construction
The Claspin-encoding DNA fragment (XhoI/XbaI fragment) of CSII-EF MCS-mAG-TEV-6xHis-Claspin-3xFlag, CSII-EF MCS-6xHis-Claspin-3xFlag or CSII-EF MCS-6xHis-Claspin-HA plasmid DNA was replaced by DNA fragments encoding portions of Claspin, amplified by PCR, to express truncated forms of Claspin.Citation19,Citation22 To express a Claspin mutant with an internal deletion, two PCR-amplified fragments (XhoI-BamHI and BamHI-XbaI or XhoI-NheI and NheI-XbaI) encoding N-terminal-proximal and C-terminal proximal segments of Claspin, respectively, were inserted at the XhoI-XbaI site of CSII-EF-MCS-6xHis-Claspin-3xFlag to replace the Claspin insert. The EcoRI-HpaI fragment of wild-type or mutant Claspin DNA from mAG-TEV-6xHis-Claspin-3xFlag was inserted at the EcoRI/SnaBI site of pMX-IP (Addgene) to construct retroviral expression vectors. The mTOR-encoding DNA fragment amplified by PCR was inserted into CSII-EF-6xHis-MCS-3xFlag3 vector DNA (ver3.4) at the BamHI site.Citation19,Citation37 Wild-type and mutant 897-1209 aa Claspin polypeptides were amplified by PCR and inserted into pET28a (+) TEV vector at NdeI site.
Analyses of apoptosis
Apoptosis was analyzed by Annexin V-FITC Apoptosis Kit (Biovision). In brief, Claspin (f/+) and Claspin (f/-) MEFs were infected with Ad-Cre or untreated for 48 h. Cells were passaged and cultured for a further 24 h. Cells were then placed in serum-free medium and were harvested at 0, 24, and 48 h. Harvested cells were labeled with propidium iodide/RNase A solution and FITC conjugated annexin V and incubated in the dark for 5 min at room temperature. The labeled cells were analyzed by FACS. Apoptosis was analyzed also by TUNEL assay, in which DNA fragmentation was analyzed by using In Situ Direct DNA Fragmentation Assay Kit (Abcam) according to the manufacturer’s protocol. Briefly, 1 × 105 Claspin(f/-) MEFs were infected with Ad-Cre or untreated for 48 h. Cells were passaged and cultured for further 24 h. Cells were then placed in serum-free medium for 48 h and harvested. Harvested cells were fixed with 1% paraformaldehyde and washed with PBS, followed by addition of 70% ice-cold ethanol for 30 min. After washing with wash buffer, cells were mixed with staining solution for 1 h at 37 °C and rinsed with the rinse buffer. After removing the rinse buffer by centrifugation, cells were re-suspended in propidium iodide/Rnase A solution and incubated in the dark for 30 min at room temperature. The FITC labeled cells were analyzed by FACS.
Senescence associated β-galactosidase activity (SA-β-gal staining)
Claspin (f/-) MEFs were infected with Ad-Cre or untreated and cultured in serum-free medium for 48 h. After addition of serum, cells were fixed at 0, 26, and 48 h by 0.25% glutaraldehyde for 5 min at room temperature. After washing with PBS, cells were stained in staining solution (5 mM potassium ferrocyanide, 40 mM citrate-Na2HPO4 (pH 6.0), 2 mM MgCl2, 150 mM NaCl and 40 mg/mL X-gal [FUJIFILM Wako Pure Chemical Corporation]) overnight at 37 °C. After washing with PBS, cells were observed under a microscope.
Antibodies
Antibodies used in this study are as follows. Anti-human Claspin, anti-mouse Claspin, anti-MCM4, and anti-MCM4 S6T7 were previously described.Citation19 Anti-LaminB (sc-6216), anti-Chk1 (sc-8408), anti-p53 (sc-99), anti-Claspin (sc-376773) and anti-MCM2 (sc-9839) were from Santa Cruz Biotechnology; anti-Flag (M185-3L) were from MBL; anti-Chk1 S345(#2341 or #2348), anti-Chk1 S317 (#2344), anti-mTor (#2983), anti-mTOR S2448 (#5536), anti-p44/42 MAPK (Erk1/2) (#4695), anti- SAPK/JNK (#9252), p38 MAPK (#8690), anti-p38 MAPK T180/Y182 (#4511), anti-p44/42 MAPK (Erk1/2) T202/Y204 (#4370), anti-SAPK/JNK T183/Y185 (#4668), anti-PDK1 (#3062), anti-PDK1 S241 (#3438), anti-PI3 kinase p110β (#3011), anti-PI3 kinase p110γ (#5405), anti-PI3 kinase p110α (#4249), anti-PI3 kinase class III (#3358), anti- p70 S6 kinase T389 (#9234), anti-p70 S6 kinase (#9202), anti-4E-BP1 T37/T46 (#2855), anti-4E-BP1 (#9644), anti-Caspase-3 (#14220), anti-Akt S473(#4058) and anti-Akt (#4691) were from Cell signaling; anti-tubulin (T5168) was from Sigma-Aldrich; anti-MCM2 S53(A300-756A) was from Bethyl; anti-p53 S37 (PAB12641) was from Abnova; anti-histone H2A.X Ser139 (05-636) and anti-histone H3 Ser10 (05-598) were from Merck Millipore; anti-Rb (GTX50459) was from GeneTex.
Cell survival measurement
Claspin (f/-) MEFs infected with Ad-Cre or untreated were cultured in serum-free medium for 48 h. After addition of serum, cells were incubated for indicated time and cell numbers were counted by staining with Trypan blue.
Knockdown of gene expression by siRNA
siRNA used are as follows. Claspin (targeting the noncoding mRNA segment) (sense: uuggccacugauuucaauutt; anti-sense: aauugaaaucaguggccaatt), Cdc7 (sense: gcagucaaagacuguggautt; anti-sense: auccacagucuuugacugctt), CK1γ1 (sense: ggcaauaagaaagagcaugtt; Anti-sense: caugcucuuucuuauugcctt), Tim (sense: gagcuaagaagccuaggggtt; anti-sense: ccccuaggcuucuuagcuctt), TopBP1 (sense: caguggagguggaguucgutt; anti-sense: acgaacuccaccuccacugtt), AND1 (sense: gguguagguaacaggacauat; anti-sense: auguccuguuaccuacaccaa), Cdc45 (sense: gaacuugaaacugcauuuctt; anti-sense: gaaaugcaguuucaaguuctt). MISSION® siRNA Universal Negative Control #1 (Merck) was used as a negative control. siRNAs were transfected into indicated mammalian cells using X-tremeGENE™ siRNA Transfection Reagent (Merck) for 48 h.
Western blotting
Protein samples were run on 5–20% precast gradient SDS-PAGE (ATTO) or 4–20% precast gradient gel (Shanghai WSHT Biotechnology Inc.) and then transferred to Hybond ECL membranes (GE Healthcare) followed by western blot analysis with the indicated antibodies. Detection was conducted with Lumi-Light PLUS Western Blotting Substrate (Roche) and images were obtained with LAS3000 (Fujifilm) or LAS4000 (GE).
BrdU incorporation
To the cells in 6 wells plates, BrdU (20 mM) was added for 20 min. Cells were harvested and were fixed at –20 °C by 75% ethanol. After washing with wash buffer (0.5% BSA in PBS), cells were treated with 2N HCl for 20 min and then with 0.1 M sodium borate (pH8.5) for 2 min, both at room temperature. Then, cells were treated with FITC-conjugated anti-BrdU antibody (BD biosciences, 51-33284X or Biolegend, 364103) for 20 min at room temperature in the dark, and further incubated with propidium iodide (25 µg/mL) and RNaseA (100 µg/mL) (Sigma-Aldrich) for 30 min at room temperature, followed by analyses with FACS.
Cell fractionation and immunoprecipitation
Cells were lysed in CSK buffer (10 mM PIPES-KOH [pH6.8], 100 mM potassium glutamate, 300 mM sucrose, 1 mM MgCl2, 1 mM EGTA, 1 mM DTT, 1 mM Na3VO4, 50 mM NaF, 0.1 mM ATP, protease inhibitor PI tablet [Roche] and 0.5 mM PMSF, 0.1% TritonX-100 and 10 units ⁄mL Benzonase [Merck Millipore]). After rotating for 60 min at 4 °C, the supernatants were recovered and were incubated with anti-Flag M2 affinity beads (Sigma) or anti-Claspin conjugated with Dynabeads™ Protein G (Thermo Fisher) for 60 min at 4 °C. The beads were washed with CSK buffer three times and proteins bound to the beads were analyzed by Western blotting.
Protein purification
Flag-tagged proteins were purified by transfection of expression plasmid DNA into 293T cells which was conducted as previously described.Citation64 Cells were lysed as above. The supernatants were mixed with anti-Flag M2 affinity beads (Sigma-Aldrich) and washed by Flag wash buffer (50 mM NaPi [pH7.5], 10% glycerol, 300 mM NaCl, 0.2 mM PMSF and PI tablet). The bound proteins were eluted by Flag elution buffer (50 mM NaPi [pH7.5], 10% glycerol, 30 mM NaCl, 200 µg/mL 3xFlag peptide [SIGMA], 0.1 mM PMSF and PI tablet).Citation41 His-tagged 897–1209 aa Claspin fragments were purified by transfection of expression plasmid DNA pET28a(+)TEV into Escherichia coli BL21-CodonPlus. Cells were lysed by lysis buffer (50 mM Na2PO4 [pH 8.0], 300 mM NaCl and 10 mM imidazole) and mixed with Ni-NAT agarose (QIAGEN) for 1 h at 4 °C. After washing by wash buffer (50 mM Na2PO4 [pH 8.0], 300 mM NaCl and 20 mM imidazole, pH 8.0), proteins were eluted by elusion buffer (50 mM Na2PO4 [pH 8.0], 300 mM NaCl and 250 mM imidazole).
Pull-down assays with purified proteins in vitro
To examine the interaction between the purified Claspin (C-terminally Flag-tagged) and PDK1 or mTOR, the recombinant wild-type or mutant Claspin proteins were incubated with purified PDK1 (Carna Bioscience) or mTOR (purified as above) in a reaction buffer (40 mM HEPES-KOH [pH 7.6], 20 mM K-glutamate, 2.5 mM MgCl2, 1 mM EGTA, 0.01% TritonX-100, 1 mM Na3VO4, 50 mM NaF, 1 mM ATP, 0.5 mM PMSF and PI tablet) at 4 °C for 1 h. Anti-Flag M2 affinity beads (for PDK1) or anti-Claspin conjugated Dynabead Protein G (for mTOR) were added and beads were recovered, washed twice with the reaction buffer. Proteins attached to the beads were analyzed by SDS-PAGE and detected by Western blotting.
ROS assay
Claspin (f/-) cells were cultured in serum-free medium for 48 h in a 48-well plate. After addition of serum, cells were cultured for indicated time and washed by Hanks’ balanced salt solution twice. After addition of DCFH-DA dye (ROS assay kit; Dojindo), cells were cultured for 30 min. After wash in Hanks’ balanced salt solution twice, cells were observed under fluorescence microscopy.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde, followed by incubation for 20 min in PBS containing 0.2% Triton X-100, which is the most popular detergent for improving the penetration of the antibody. After washing by PBS, cells were incubated in PBST/1% BSA for 1 h, followed by incubation with anti-Claspin and anti-mTOR antibodies in PBST/1% BSA overnight at 4 °C. After washing, cells were incubated with anti-mouse IgG (Alexa Fluor® 488) (Invitrogen) and anti-rabbit IgG (Alexa Fluor® 647) (Invitrogen) secondary antibodies in 1% BSA for 1 h at room temperature in the dark. After washing, cells were mounted and observed under fluorescence microscope.
Mass spectrometry analyses of Claspin
The 293T cells (15 cm dish, five plates) were cultured in serum-containing or serum-free medium for 24 h. Cells were lysed with RIPA buffer (50 mM Tris-HCl [pH8.0], 150 mM NaCl, 0.5% w/v sodium deoxycholate, 1.0% NP-40 and 0.1% SDS) containing 1 mM DTT, 1 mM Na3VO4, 50 mM NaF, 0.1 mM ATP, 0.5 mM PMSF and PI tablet. The supernatants were mixed with anti-Claspin antibody conjugated to Dynabead protein G at 4 °C for 1 h. After washes with PBS containing 0.01% Tween-20, the proteins on beads were separated on 4–20% gradient gel and stained by CBB. Claspin protein band was excised from the gel, digested by Trypsin and phospho-threonines or phosphoserines were analyzed by mass spectroscopy.
Pull-down assays with CKBD peptides in vitro
To examine the interactions between the phosphorylated CKBD peptide of Claspin and proteins involved in the PI3K-PDK1-mTOR pathway, 200 ng biotin-tagged phosphorylated CKBD peptide of Claspin, treated with λPPase or untreated, were incubated with purified proteins in a reaction buffer (40 mM HEPES-KOH [pH 7.6], 20 mM K-glutamate, 2.5 mM MgCl2, 1 mM EGTA, 0.01% Triton X-100, 1 mM Na3VO4, 50 mM NaF, 1 mM ATP, 0.5 mM PMSF and PI tablet) at 4 °C for 1 h. Streptavidin beads were added and incubated at 4 °C for 1 h, then washed twice with the reaction buffer. Proteins attached to the beads were analyzed by SDS-PAGE and detected by Western blotting.
In vitro kinase assays of Claspin-derive polypeptides
DNA fragments coding wild-type, ST27/A or ST27/E (all the serine/threonine in the 903-1120 aa segment changed to alanine or glutamic acid, respectively) mutant of Claspin polypeptides (897-1209 aa) were cloned at NdeI site of pET28a(+)TEV vector, and proteins were purified by Ni-NTA agarose. The purified Claspin-derive polypeptides were incubated with the purified PDK1, mTOR, PIK3CB or PIK3C3 proteins in the kinase reaction buffer (40 mM HEPES-KOH [pH7.6], 5 mM MgCl2, 0.5 mM EGTA, 0.5 mM EDTA, 1 mM Na3VO4, 1 mM NaF, 2 mM DTT, 10 μM ATP, and 1 μCi [γ-ATP]) for 1 h at 37 °C. After addition of one-fourth volume of 5× sample buffer, the reaction mixtures were heated at 95 °C for 1 min and analyzed by SDS-PAGE, followed by CBB staining and autoradiogram, to detect phosphorylation.
Effects of kinase on Claspin interaction with PDK1 by pull-down assay
The purified Claspin-derived polypeptides (897–1209 aa) were incubated with the PDK1, PIK3CB, Cdc7 or CK1γ1 in the kinase reaction buffer (40 mM HEPES-KOH [pH7.6], 5 mM MgCl2, 0.5 mM EGTA, 0.5 mM EDTA, 1 mM Na3VO4, 1 mM NaF, 2 mM DTT, 10 μM ATP, and 1 μCi [γ-ATP]) for 1 h at 37 °C. Ni-NTA agarose were added to the reaction mixtures and the collected beads were washed with Flag wash buffer as described above. Samples were analyzed by Western blotting with indicated antibodies.
Effects of kinase inhibitors on cellular protein interactions by pull-down assay
Flag-tagged Claspin were overexpressed in 293T cells for 24 h and then treated with DMSO (control), 2 µM PI-103, 5 µM GSK2334470 or 2.5 µM rapamycin for 12 h. Cells were lysed with CSK buffer as above. The supernatants were mixed with anti-Flag M2 affinity beads and the collected beads were washed with Flag wash buffer as described above. Samples were analyzed by Western blotting with indicated antibodies.
Effects of kinase inhibitors on cell viability after serum starvation and during release from serum starvation
Three sets of 293T cells were transfected with siControl or siClaspin for 24 h in serum-free medium for 24 h. One set was harvested at 24 h after serum starvation. The remaining two sets were released into growth by addition of serum for 12 h or 24 h. Asynchronously growing 293T cells were also transfected with siControl or siClaspin for 24 h. Inhibitors or DMSO (control) were added during the last 12 h before the harvest. Inhibitors added and their concentrations were as follows: Wortmannin (FUJIFILM Wako Pure Chemical Corporation), 2 µM; PI-103 (Cayman Chemical Company), 2 µM; Flavopiridol (Selleck), 10 µM; UCN-01 (Cayman Chemical Company), 1 µM; VE-821(Sigma-Aldrich), 10 µM; GSK2334470 (Selleck), 5 µM; XL413(AdooQ Bioscience), 10 µM; Torkinib (Selleck), 5 µM; rapamycin (Medchemexpress), 2.5 µM and MIRIN (Sigma-Aldrich), 10 µM. Cells were observed under a microscope.
Statistical analyses
Western blotting and FACS data were generated in three independent experiments in most cases and in at two independent experiments in some cases (indicated in the figure legends). Error bars represent the mean ± SD values calculated from three independent replicate experiments.
RNA sequencing
RNA was isolated from Claspin (f/-) MEFs grown under three different conditions (grown asynchronously, grown in serum-free medium for 24 h, and grown in serum-free medium for 24 h and released into growth by addition of serum for 14 h) by using RNA purification kit (QIAGEN). Purified RNAs were sequenced by GENEWIZ. Raw data were analyzed by CLC Genomics Workbench (Qiagen).
CONCLUSIONS
We have identified a novel role of Claspin in activation of the PI3K-PDK1-mTOR during growth restart from serum starvation. The PI3K-Claspin-PDK in serum-induced signal transmission may parallel the ATR-Claspin-Chk1 in replication stress checkpoint. PDK1 binds to phosphorylated CKBD of Claspin, like Chk1, and phosphorylates Claspin, which in turn promotes recruitment of mTOR. Claspin may serve as a protein hub for activation of the PI3K-Claspin-PDK (). Our findings also point to an intriguing possibility that Claspin is a general mediator that links upstream PIKK to downstream effectors in varied environmental settings.
AUTHOR CONTRIBUTIONS
C-CY and HM conceived the research plans. C-CY conducted most of the experiments. HM conducted some of the plasmid constructions for expression of recombinant proteins used in this study. C-CY and HM wrote the paper.
ACKNOWLEDGMENTS
We thank Mayumi Shindo of Protein Analyses Laboratory of our institute for analyses of phosphorylation of the Claspin and Naoko Kakusho and Rino Fukatsu for excellent technical assistance. We also thank the members of our laboratory for useful discussion.
DATA AVAILABILITY STATEMENT
Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding author.
Additional information
Funding
REFERENCES
- Branzei D, Foiani M. The checkpoint response to replication stress. DNA Repair. 2009;8:1038–1046. doi:10.1016/j.dnarep.2009.04.014.
- Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21:4129–4139. doi:10.1128/MCB.21.13.4129-4139.2001.
- Lindsey-Boltz LA, Serçin Ö, Choi JH, Sancar A. Reconstitution of human claspin-mediated phosphorylation of Chk1 by the ATR (ataxia telangiectasia-mutated and Rad3-related) checkpoint kinase. J Biol Chem. 2009;284:33107–33114. doi:10.1074/jbc.M109.064485.
- Kumagai A, Dunphy WG. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol Cell. 2000;6:839–849. doi:10.1016/S1097-2765(05)00092-4.
- Tanaka K, Russell P. Mrc1 channels the DNA replication arrest signal to checkpoint kinase Cds1. Nat Cell Biol. 2001;3:966–972. doi:10.1038/ncb1101-966.
- Alcasabas AA, Osborn AJ, Bachant J, Hu F, Werler PJH, Bousset K, Furuya K, Diffley JF, Carr AM, Elledge SJ. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat Cell Biol. 2001;3:958–965. doi:10.1038/ncb1101-958.
- Christiano C, Chini S, Chen J. Human claspin is required for replication checkpoint control. J Biol Chem. 2003;278:30057–30062. doi:10.1074/jbc.M301136200.
- Osborn AJ, Elledge SJ. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 2003;17:1755–1767. doi:10.1101/gad.1098303.
- Hae YY, Jeong SY, Dunphy WG. Site-specific phosphorylation of a checkpoint mediator protein controls its responses to different DNA structures. Genes Dev. 2006;20:772–783. doi:10.1101/gad.1398806.
- Kim JM, Kakusho N, Yamada M, Kanoh Y, Takemoto N, Masai H. Cdc7 kinase mediates Claspin phosphorylation in DNA replication checkpoint. Oncogene. 2008;29;27(24):3475–3482. doi:10.1038/sj.onc.1210994.
- Kumagai A, Dunphy WG. Repeated phosphopeptide motifts in claspin mediate the regulated binding of Chk1. Nat Cell Biol. 2003;5:161–165. doi:10.1038/ncb921.
- Chini CCS, Chen J. Repeated phosphopeptide motifs in human claspin are phosphorylated by Chk1 and mediate claspin function. J Biol Chem. 2006;281:33276–33282. doi:10.1074/jbc.M604373200.
- Meng Z, Capalbo L, Glover DM, Dunphy WG. Role for casein kinase 1 in the phosphorylation of claspin on critical residues necessary for the activation of Chk1. Mol Biol Cell. 2011;22:2834–2847. doi:10.1091/mbc.e11-01-0048.
- Yang CC, Kato H, Shindo M, Masai H. Cdc7 activates replication checkpoint by phosphorylating the chk1 binding domain of claspin in human cells. Elife. 2019;8:e50796. doi:10.7554/eLife.50796.
- Szyjka SJ, Viggiani CJ, Aparicio OM. Mrc1 is required for normal progression of replication forks throughout chromatin in S. cerevisiae. Mol Cell. 2005;19:691–697. doi:10.1016/j.molcel.2005.06.037.
- Tourrière H, Versini G, Cordón-Preciado V, Alabert C, Pasero P. Mrc1 and Tof1 promote replication fork progression and recovery independently of Rad53. Mol Cell. 2005;19:699–706. doi:10.1016/j.molcel.2005.07.028.
- Yeeles JTP, Janska A, Early A, Diffley JFX. How the eukaryotic replisome achieves rapid and efficient DNA replication. Mol Cell. 2017;65:105–116. doi:10.1016/j.molcel.2016.11.017.
- Petermann E, Helleday T, Caldecott KW. Claspin promotes normal replication fork rates in human cells. Mol Biol Cell. 2008;19:2373–2378. doi:10.1091/mbc.e07-10-1035.
- Yang CC, Suzuki M, Yamakawa S, Uno S, Ishii A, Yamazaki S, Fukatsu R, Fujisawa R, Sakimura K, Tsurimoto T, et al. Claspin recruits Cdc7 kinase for initiation of DNA replication in human cells. Nat Commun. 2016;7:12135. doi:10.1038/ncomms12135.
- Masai H, Yang CC, Matsumoto S. Mrc1/Claspin: a new role for regulation of origin firing. Curr Genet. 2017;63(5):813–818. doi:10.1007/s00294-017-0690-y.
- Hsiao, HW., Yang, CC, Masai, H. Roles of Claspin in regulation of DNA replication, replication stress responses and oncogenesis in human cells. Genome Instab Dis. 2021;2:263–280. doi:10.1007/s42764-021-00049-8.
- Uno S, Masai H. Efficient expression and purification of human replication fork-stabilizing factor, Claspin, from mammalian cells: DNA-binding activity and novel protein interactions. Genes Cells. 2011;16(8):842–856. doi:10.1111/j.1365-2443.2011.01535.x.
- Hayano M, Kanoh Y, Matsumoto S, Masai H. Mrc1 marks early-firing origins and coordinates timing and efficiency of initiation in fission yeast. Mol Cell Biol. 2011;31(12):2380–91. doi:10.1128/MCB.01239-10.
- Duch A, Felipe-Abrio I, Barroso S, Yaakov G, García-Rubio M, Aguilera A, de Nadal E, Posas F. Coordinated control of replication and transcription by a SAPK protects genomic integrity. Nature. 2013;493:116–121. doi:10.1038/nature11675.
- Duch A, Canal B, Barroso SI, García-Rubio M, Seisenbacher G, Aguilera A, de Nadal E, Posas F. Multiple signaling kinases target Mrc1 to prevent genomic instability triggered by transcription-replication conflicts. Nat Commun. 2018;9:379. doi:10.1038/s41467-017-02756-x.
- de Nadal E, Posas F. Osmostress-induced gene expression – a model to understand how stress-activated protein kinases (SAPKs) regulate transcription. FEBS J. 2015;282: 3275–3285. doi:10.1111/febs.13323.
- Cabrera E, Hernández-Pérez S, Koundrioukoff S, Debatisse M, Kim D, Smolka MB, Freire R, Gillespie DA. PERK inhibits DNA replication during the unfolded protein response via Claspin and Chk1. Oncogene. 2017;36:678–686. doi:10.1038/onc.2016.239.
- McDonald NQ, Hendrickson WA. A structural superfamily of growth factors containing a cystine knot motif. Cell. 1993;73: 421–424. doi:10.1016/0092-8674(93)90127-C.
- Plotnikov AN, Hubbard SR, Schlessinger J, Mohammadi M. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell. 2000;101:413–424. doi:10.1016/S0092-8674(00)80851-X.
- Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, Franklin RA, McCubrey JA. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17: 590–603. doi:10.1038/sj.leu.2402824.
- Toker A, Cantley LC. Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature. 1997;387: 673–676. doi:10.1038/42648.
- Vanhaesebroeck B, Leevers SJ, Panayotou G, Waterfield MD, Waterfield MD. Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends Biochem Sci. 1997;22: 267–272. doi:10.1016/S0968-0004(97)01061-X.
- Leevers SJ, Vanhaesebroeck B, Waterfield MD. Signalling through phosphoinositide 3-kinases: the lipids take centre stage. Curr Opin Cell Biol. 1999;11(2), 219–225. doi:10.1016/s0955-0674(99)80029-5.
- Williams MR, Arthur JSC, Balendran A, Van der Kaay J, Poli V, Cohen P, Alessi DR. The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr Biol. 2000;10:439–448. doi:10.1016/S0960-9822(00)00441-3.
- Toker A, Newton AC. Cellular signaling: pivoting around PDK-1. Cell. 2000;103: 185–188. doi:10.1016/S0092-8674(00)00110-0.
- Belham C, Shilan W, Avruch J. Intracellular signalling: PDK1 – a kinase at the hub of things. Curr Biol. 1999;9:R93–R96. doi:10.1016/S0960-9822(99)80058-X.
- Ma Y, Vassetzky Y, Dokudovskaya S. mTORC1 pathway in DNA damage response. Biochim Biophys Acta - Mol Cell Res. 2018;1865:1293–1311. doi:10.1016/j.bbamcr.2018.06.011.
- Navé BT, Ouwens DM, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344:427–431. doi:10.1042/bj3440427.
- Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science. 2001;294:1942–1945. doi:10.1126/science.1066015.
- Pullen N, Thomas G. The modular phosphorylation and activation of p70(s6k). FEBS Lett. 1997;410:78–82. doi:10.1016/S0014-5793(97)00323-2.
- Dufner A, Thomas G. Ribosomal S6 kinase signaling and the control of translation. Exp Cell Res. 1999;253:100–109. doi:10.1006/excr.1999.4683.
- Jewell JL, Guan KL. Nutrient signaling to mTOR and cell growth. Trends Biochem Sci. 2013;38:233–242. doi:10.1016/j.tibs.2013.01.004.
- Budanov AV., Karin M. p53 target genes Sestrin1 and Sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–460. doi:10.1016/j.cell.2008.06.028.
- Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ. The regulation of AMPK β1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67:3043–3053. doi:10.1158/0008-5472.CAN-06-4149.
- Budanov AV., Sablina AA, Feinstein E, Koonin EV., Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science. 2004;304:596–600. doi:10.1126/science.1095569.
- Zhang X, Shu L, Hosoi H, Gopal Murti K, Houghton PJ. Predominant nuclear localization of mammalian target of rapamycin in normal and malignant cells in culture. J Biol Chem. 2002;277:28127–28134. doi:10.1074/jbc.M202625200.
- Devarenne TP, Ekengren SK, Pedley KF, Martin GB. Adi3 is a Pdk1-interacting AGC kinase that negatively regulates plant cell death. EMBO J. 2006;25:255–265. doi:10.1038/sj.emboj.7600910.
- Romero-Pozuelo J, Figlia G, Kaya O, Martin-Villalba A, Teleman AA. Cdk4 and Cdk6 couple the cell-cycle machinery to cell growth via mTORC1. Cell Rep. 2020;31: 107504. doi:10.1016/j.celrep.2020.03.068.
- Kumar A, Redondo-Munoz J, Perez-Garcia V, Cortes I, Chagoyen M, Carrera AC. Nuclear but not cytosolic phosphoinositide 3-kinase beta has an essential function in cell survival. Mol Cell Biol. 2011;31:2122–2133. doi:10.1128/MCB.01313-10.
- Marqués M, Kumar A, Poveda AM, Zuluaga S, Hernández C, Jackson S, Pasero P, Carrera AC. Specific function of phosphoinositide 3-kinase beta in the control of DNA replication. Proc Natl Acad Sci U S A. 2009;106:7525–7530. doi:10.1073/pnas.0812000106.
- Budanov AV. The role of tumor suppressor p53 in the antioxidant defense and metabolism. Subcell Biochem. 2014;85:337–358. doi:10.1007/978-94-017-9211-0_18.
- Czarny P, Pawlowska E, Bialkowska-Warzecha J, Kaarniranta K, Blasiak J. Autophagy in DNA damage response. Int J Mol Sci. 2015;16: 2641–2662. doi:10.3390/ijms16022641.
- Hasty P, Sharp ZD, Curiel TJ, Campisi J. mTORC1 and p53: clash of the gods? Cell Cycle. 2013;12: 20–25. doi:10.4161/cc.22912.
- Liebl MC, Hofmann TG. Cell fate regulation upon DNA damage: p53 serine 46 kinases pave the cell death road. BioEssays. 2019;41:e1900127. doi:10.1002/bies.201900127.
- Lai KP, Leong WF, Chau JFL, Jia D, Zeng L, Liu H, He L, Hao A, Zhang H, Meek D, et al. S6K1 is a multifaceted regulator of Mdm2 that connects nutrient status and DNA damage response. EMBO J. 2010;29:2994–3006. doi:10.1038/emboj.2010.166.
- He Z, Simon HU. A novel link between p53 and ROS. Cell Cycle. 2013;12:201–202. doi:10.4161/cc.23418.
- Achanta G, Huang P. Role of p53 in sensing oxidative DNA damage in response to reactive oxygen species-generating agents. Cancer Res. 2004;64:6233–6239. doi:10.1158/0008-5472.CAN-04-0494.
- Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169:361–371. doi:10.1016/j.cell.2017.03.035.
- Wang X, Proud CG. The mTOR pathway in the control of protein synthesis. Physiology. 2006;21:362–369. doi:10.1152/physiol.00024.2006.
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi:10.1101/gad.1212704.
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi:10.1038/nrm2672.
- Muñoz IM, Hain K, Déclais AC, Gardiner M, Toh GW, Sanchez-Pulido L, Heuckmann JM, Toth R, Macartney T, Eppink B, et al. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol Cell. 2009;35:116–127. doi:10.1016/j.molcel.2009.06.020.
- Tesmer JJG. Chapter 4. Structure and function of regulator of G protein signaling homology domains. Prog Mol Biol Transl Sci. 2009;86:75–113.
- Uno S, You Z, Masai H. Purification of replication factors using insect and mammalian cell expression systems. Methods. 2012;57:214–21. doi:10.1016/j.ymeth.2012.06.016.