Abstract
We have linked phylogenetics with population genetics in order to investigate the effects of development mode on the genetic diversity and population structure of two southern South American marine gastropod species, Crepipatella dilatata and C. fecunda. DNA sequences were obtained for the mitochondrial gene cytochrome oxidase (COI). We have found that the mean level of genetic diversity was significantly greater in C. dilatata than in C. fecunda, the latter having the highest number of shared haplotypes. The AMOVA analysis shows that C. dilatata has a marked population genetic structure, with 56.86% of the observed genetic variation explained by the variation between populations, in contrast to the genetic homogeneity exhibited by C. fecunda in which most of the variance (97.33%) is explained by the intrapopulational variation. For C. dilatata the multimodal curve obtained is consistent with a long-term stationary population model, whereas in the case of C. fecunda, the mismatch distribution shows a significant fit to a population growth model. Both, the level of genetic differentiation among populations as well as the patterns of genetic structure, correlate well with the larval developmental mode. The role of the environment, selection and historical events on the level of genetic differentiation and the population genetic structure are discussed for both species.
Introduction
The mode of development is a key trait in the life history of many marine invertebrates, and it is considered a strong predictor of the dispersal potential and the consequent genetic structuring of the populations (Levin Citation2006). As planktonic larvae can remain suspended in the water column at the mercy of ocean currents for weeks or months, it is commonly supposed that benthic species with planktonic larvae have higher levels of dispersal and lower degrees of genetic structuring than species with direct development (Kelly & Palumbi Citation2010). Comparative studies which have examined the population structure of echinoderms (Arndt & Smith Citation1998; Iuri et al. Citation2007), bryozoans (Watts & Thorpe Citation2006), crustaceans (Duffy Citation1993), corals (Hellberg Citation1996; Ayre & Hughes Citation2000), and marine gastropods (Janson Citation1987; Wilke & Davis Citation2000; Collin Citation2001) support the prediction that species with pelagic larvae show low degrees of population structure and genetic differentiation across populations separated by hundreds of kilometres.
Marine gastropods of the genus Crepipatella (Lesson, 1830) are among the most well-studied species of the family Calyptraeidae (Gallardo Citation1977, 1979; Collin Citation2003; Véliz et al. Citation2003; Chaparro et al. Citation2005; Collin et al. Citation2007), and recent studies indicate that there are five valid species: C. dilatata (Lamarck, 1822), C. capensis (Quoy & Gaimard, 1833), C. dorsata (Broderip, 1834), C. lingulata (Gould, 1846), and C. fecunda (Gallardo, Citation1979). Two of these species, C. fecunda and C. dilatata, inhabit rocky shores along the Chilean and Argentinean coasts.
From a morphological point of view, C. dilatata and C. fecunda are indistinguishable as adults, being recognized only on the basis of some key traits in their development (Gallardo Citation1979; Véliz et al. Citation2003, Collin et al. Citation2007). C. dilatata exhibits intracapsular development with embryos that consume uncleaved nurse eggs, and hatch as crawling juveniles with a shell length of about 1100–1200 μm. On the other hand, C. fecunda exhibits pelagic development, embryos do not consume nurse eggs, and they hatch as planktotrophic larvae at a size of 400–500 μm (Gallardo Citation1977, Citation1979; Chaparro et al. Citation2002; Véliz et al. Citation2003; Collin et al. Citation2007).
On the southeastern Pacific coast the distributional range of C. dilatata extends from 12° 06′S (Isla San Lorenzo, Perú) to 55° 00′S (Tierra del Fuego, Chile), whereas the geographical range of C. fecunda extends from 11°47′S (Ancón, Perú) to 45° 43′S (Fiordo de Quitralco, Chile). At some locations within this range, such as Coquimbo (29°59′S), Chinquihue (41°31′S), and Quempillén (41°52′S) C. dilatata and C. fecunda are sympatric, a situation in which C. dilatata forms less abundant populations (Gallardo Citation1979). C. dilatata also occurs on the southwestern Atlantic coast at Puerto Madryn (Argentina).
From the molecular point of view (mitochondrial DNA sequences), both species present < 2% of genetic divergence (Collin Citation2002, Citation2003). In the same line, analyses of 26 allozyme loci (Véliz et al. Citation2003) showed that both species are closely related (Nei's genetic distance < 0.25). Phylogenetic reconstructions show C. fecunda within a paraphyletic group of C. dilatata, suggesting that C. dilatata is the extant ancestor of C. fecunda (Collin et al. Citation2007). Further, the paraphyly of C. dilatata by all three phylogenetic methods is pointed out by Collin et al. (Citation2007) as the expected population structure from a direct-developing species.
In the present study we have evaluated the variation of mitochondrial cytochrome oxidase I (COI) sequences in order to examine the genetic structure of C. dilatata and C. fecunda in relation to their development mode. Molecular markers such as the mitochondrial DNA (mtDNA) are commonly used to study the levels of genetic diversity, population structure and make inferences about gene flow in aquatic invertebrates (Zickovich & Bohonak Citation2007). In fact, some population genetic statistics correlate predictably with dispersal ability across a wide variety of taxa (Peterson & Denno Citation1998; Kelly & Palumbi Citation2010). With this in mind, our preliminary hypothesis predicts that populations of C. fecunda will exhibit lower degrees of population structuring and genetic differentiation than populations of C. dilatata.
Materials and methods
Biological sampling
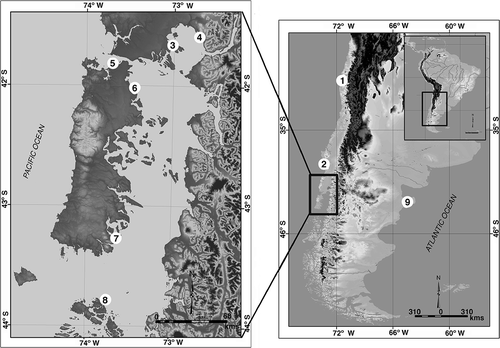
Specimens of C. fecunda and C. dilatata were surveyed along the Chilean Pacific coast; in addition, one population of C. dilatata was collected at Puerto Madryn, on the Atlantic coast (; ). Specimens were identified according to the developmental mode observed in their capsules (Gallardo Citation1979). Approximately 500 mg of foot muscle tissue of each specimen was fixed and preserved in absolute ethanol. Furthermore, 34 additional sequences available from GenBank were included in our genetic data set.
Table I. Genetic diversity estimates of C. dilatata and C. fecunda. N refers to the number of individuals sampled, S is the number of segregating sites, H is the number of haplotypes, and for all populations refers to unique haplotypes. Hd is the haplotype diversity and π is the nucleotidic diversity, both with standard deviation in parenthesis. *Significant values at p < 0.05. NA, not applicable
Figure 1. Map of the Chilean and Argentinean coasts showing the sampling locations of animals used in this study. 1, Coquimbo; 2, Corral; 3, Chinquihue; 4, Metri; 5, Quempillén; 6, Aucho; 7, Yaldad; 8, Melinka; 9, Puerto Madryn. Ancon and Santa María locations from Perú are not shown, as such sequences were obtained from GenBank.
Genetic techniques
Whole genomic DNA was extracted from tissue samples using standard protocols of chemical digestion followed by phenol/chloroform extraction (Sambrook et al. Citation1989). Polymerase chain reaction (PCR) amplifications were performed using 50–100 ng of genomic DNA and 0.2 U of DNA polymerase (Invitrogen) per 50 μl of reaction volume. Amplification and sequencing of the COI fragments were performed using the primers LCO1490 and HCO 2198 (Folmer et al. Citation1994). The PCR cycling profile was as follows: an initial denaturation at 94°C for 5 min, followed by 35 cycles at 92°C for 50 s, 52°C for 45 s, and 72°C for 1 min, and ending with chain extension at 72°C for 7 min. The PCR products were visualized on an ethidium bromide-stained agarose gel (1%) using UV illumination. Purified PCR products were sequenced at Macrogen Inc. (Seoul, Korea). Sequences were submitted to the National Center for Biotechnology Information (NCBI) database, using SEQUIN (http://www.ncbi.nlm.nih.gov/Sequin) under the Accession Numbers JF705954-JF705984. Comparisons of the obtained nucleotide sequences with those of C. fecunda and C. dilatata available in the GenBank database were performed using the online BLAST program. All nucleotide sequences were aligned using MAFFT 6 (Katoh et al. Citation2005) using the iterative method of global pairwise alignment (G-INS-i), and default settings were chosen for all the parameters involved.
Phylogenetic analyses
A phylogenetic analysis was performed by Neighbour Joining (NJ) based on K2P distance. Reliabilities of phylogenetic relationships were evaluated using nonparametric bootstrap analysis (Felsenstein Citation1985) with 1000 replicates. To confirm the obtained tree from NJ analysis, another phylogenetic tree was calculated using MrBayes v3.0b3 (Huelsenbeck & Ronquist Citation2003). In this analysis, four incrementally heated Markov Chains were run for 10,000,000 generations (ngen = 10,000,000), sampling every 100 generations (samplefreq = 100), where 2500 samples were discarded (burnin = 2500). COI sequence of C. capensis (AF546053) was used to root the trees.
Haplotype networks
Intraspecific gene genealogies for each species of Crepipatella were inferred using the median-joining network algorithm implemented in NETWORK v4.51 (Bandelt et al. Citation1999). The median-joining method uses a maximum parsimony approach to search for all the shortest phylogenetic trees for a given data set. To construct the network, we weighed transversions twice as high as transitions (Bandelt et al. Citation1999). After generating the initial network, we used the MP option to remove excessive links and median vectors and construct the final median-joining network. The tolerance parameter epsilon was set to 0, and haplotype frequencies were converted into proportional areas in the graphical output.
Population genetic analyses
Haplotype and nucleotide diversity (Nei Citation1987), as well as their standard deviations, were estimated with DnaSP 5.0 (Librado & Rozas Citation2009). The possibility of saturation in the rate of base substitutions at the COI gene was assessed by the method of Xia et al. (Citation2003) using the DAMBE program (Xia & Xie Citation2001). Demographic patterns were examined with two approaches. First, we evaluated whether the COI fragment evolved under strict neutrality. For this, we used Tajima's D (Tajima Citation1989), Fu & Li's D (Fu & Li Citation1993), and R2 (Ramos-Onsins & Rozas Citation2002) tests. Significant values of any of these may indicate that the populations are not evolving in a neutral manner (i.e. they are not in mutation-drift equilibrium), or that they were previously subdivided and/or have experienced past population growth. The second hypothesis of historical demographic expansions was investigated by the examination of ‘mismatch distribution’, that is the frequency distributions of pairwise differences between sequences (Rogers & Harpending Citation1992; Excoffier Citation2004). Mismatch distributions can be used to test the hypotheses concerning population demographic history and selection. Under this approach, the smooth, unimodal distributions that typically result from expanding populations can be readily distinguished from the ragged, multimodal distributions that results from long-term stationary populations by means of the measure of ‘raggedness’ of the distribution (Rogers & Harpending Citation1992). Confidence intervals for these statistics were estimated by means of computer simulations using the coalescence algorithm as implemented in DnaSP 5.0.
In order to explore patterns of global genetic structure between sampling locations of C. fecunda and C. dilatata we performed a Spatial Analysis of Molecular Variance (SAMOVA) using the program SAMOVA 1.0 (Dupanloup et al. Citation2002). SAMOVA is a nonphylogenetic method based on the fixation index Fct, in which the geographical information of each population is combined with an Analysis of Molecular Variance (AMOVA; Excoffier et al. Citation1992), and then a simulated annealing procedure maximizes the total amount of molecular variance due to differences between groups of geographically homogeneous populations. We performed the SAMOVA analysis based on 200 simulated annealing steps for k = n–1 partitions, where k was the number of genetically defined units (i.e. groups), and n was the number of populations included in the analysis. This was followed by an AMOVA in Arlequín V 3.11 (Schneider et al. Citation2006) to compute F ST values, and explore in more detail the partitioning of the genetic variance among the maximally differentiated groups identified by SAMOVA.
In order to test the hypothesis of isolation by distance (IBD), estimates of pairwise F ST were plotted against geographical distance using Isolation by Distance Web Service version 3.14 (Jensen et al. Citation2005) with 30,000 randomizations. This web service runs a Mantel's test and calculates correlation between pairwise F ST and geographic distances using a reduced major axis regression. Geographic distances consisted of the shortest path by sea between any two populations, calculated using Google Earth version 5.1.35.
Results
Descriptive analysis of sequence data
Individuals from different geographic locations were clearly identified through the observation of ovicapsules and classified according to developmental modes described by Gallardo (Citation1977). Haplotype diversity of C. dilatata ranged between 0.159 and 0.807, whereas haplotype diversity of C. fecunda ranged between 0.571 and 0.825 (). The ratio of the unique haplotypes relative to the total individuals sampled by species was 0.222 for C. dilatata and 0.209 for C. fecunda. The highest Hd value was for C. fecunda from Metri (0.825), while the lowest was from Quempillén (0.159). In C. dilatata a total of 14 distinct COI haplotypes were identified from the 63 individuals examined in this study (). Of these, six haplotypes were sampled more than once, while the remaining occurred as singletons. In C. fecunda, a total of 18 distinct COI haplotypes were identified from the 86 individuals examined (). Of these, seven haplotypes were sampled more than once, while the remaining occurred as singletons. Saturation plots revealed that transitions and transversions were not saturated.
Table II. Distribution of haplotypes per locality of C. dilatata.
Table III. Distribution of haplotypes per locality of C. fecunda
Phylogenetic reconstruction
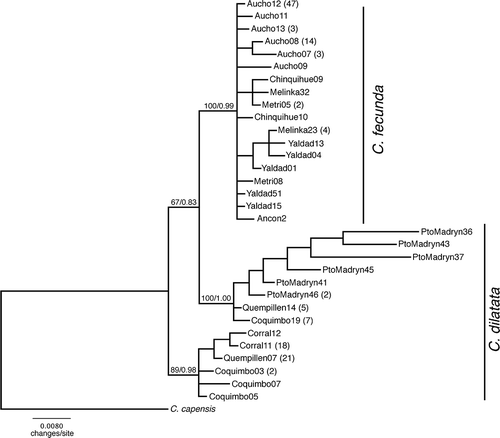
Phylogenetic analysis of C. fecunda and C. dilatata sequences produced identical branching patterns by both different methods: the Neighbour Joining and the Bayesian inference. However, the Bayesian tree was considered the most accurate tree out of two calculated trees, so we chose it as a reference tree in this study (). A clade of C. fecunda was recovered by both methods with high bootstrap (100%) and posterior probability (0.99) values. C. dilatata was recovered as a paraphyletic group in which two clades with high bootstrap and posterior probability values can be distinguished, one composed of populations from Pacific coasts, and the other one composed mostly of Puerto Madryn Atlantic coasts.
Figure 2. Bayesian tree representing the relationships between C. fecunda and C. dilatata populations. Numbers above the branches represent bootstrap support value and Bayesian posterior probabilities. Numbers in parenthesis indicate the number of individuals sharing the respective sequence.
Haplotype networks
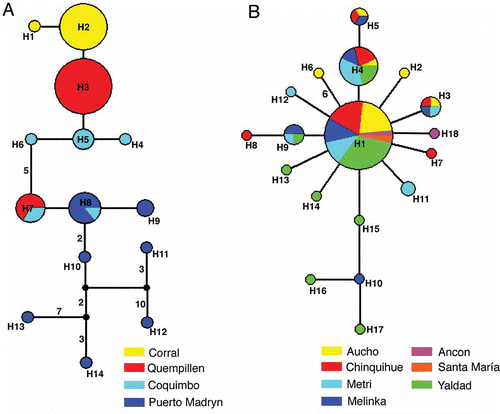
The median-joining network representing the relationships between haplotypes of C. dilatata is shown in . In most cases, individuals from the same sampling site were clustered together. The exception was observed for Puerto Madryn haplotypes, where they were mostly singletons. On the other hand, the median-joining network for C. fecunda haplotypes () lacked the distinct clustering of haplotypes from a single sample location, and the most common haplotype (H1) was the central hub in this configuration. As a result, we did not obtain population structure, but rather a high genetic homogeneity among the populations of C. fecunda, in contrast to that obtained for C. dilatata.
Figure 3. Median-joining network representing the relationships among haplotypes of Crepipatella dilatata (A) and Crepipatella fecunda (B). Small black circles indicate median vectors (Bandelt et al. Citation1999). Circle size is proportional to haplotype frequencies, with the smaller circle representing one sample. Numbers at the branches represent the number of mutations separating the haplotypes if greater than one.
Population genetic analysis
Neutrality tests applied to the data sets showed that Tajima's D, Fu and Li's D, and R2 statistic values were all nonsignificant for C. dilatata except for the Coquimbo population (). In the case of C. fecunda, Tajima's D, Fu and Li's D, and R2 statistics values were all consistent with the prediction for an expanding population, however these values were nonsignificant for each population. In contrast, pooled samples of C. fecunda produced significantly negative Fu & Li's D (−2.9516, p < 0.05) and Tajima's D (−1.9800, p < 0.05) values, suggesting that population stasis/neutrality should be rejected and indicates an excess of recent mutations that could be explained by a recent population expansion model.
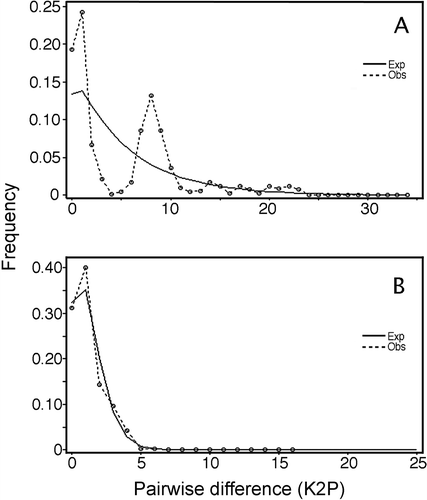
Results of mismatch distribution of haplotypes for both species are shown in . A multimodal distribution of pairwise differences which is commonly inferred for long-term stationary populations was observed in the case of C. dilatata (), contrasting with the smooth, unimodal distribution for C. fecunda () consistent with a sudden population expanding model (Harpending Citation1994). In the latter case, the Harpending raggedness was low (r = 0.09455, p < 0.05) indicating a significant fit between the observed and expected distribution, providing further evidence for a recent population expansion model for this species.
Figure 4. Mismatch distributions observed (Obs) for (A) Crepipatella dilatata and (B) Crepipatella fecunda. The expected (Exp) frequency is based on a sudden population expansion model. Goodness-of-fit of the observed versus the theoretical mismatch distribution was tested using the sum of squared deviations (SSD).
Results of the spatial variance analysis, SAMOVA for C. dilatata are summarized in . When k = 2, 61.70% of the molecular variance was due to differences between groups and 10.63% due to differences within both groups, resulting in a value of Fct = 0.617, significant at p < 0.05. Along the same lines, when k = 3, 58.85% of the molecular variance was due to differences between groups and only 7.63% due to differences within groups, resulting in a value of Fct= 0.588, significant at p < 0.05. On the other hand, SAMOVA for the C. fecunda population samples () indicated that when more groups were defined (k = 2–6), the Fct value increased but some groups began to consist of a single population. In all cases, no genetic differentiation between groups was found (Fct= 0.04066–0.18935, p < 0.05).
Table IV. Summary of SAMOVA for C. dilatata. The k-values represent number of groups hypothesized. *p-value is < 0.05
Table V. Summary of SAMOVA for C. fecunda. The k-values represent number of groups hypothesized. p-value is < 0.05
The AMOVA analysis found that most of the observed genetic variation in C. dilatata occurred between populations (F ST = 0.568, p < 0.001), while variation within populations explained 43.14% of the variance (). In contrast, nonsignificant genetic differentiation between populations was found for C. fecunda, where most of the variance (97.33%) was explained by the intrapopulational variation. Isolation by distance analyses revealed a non significant relationship between genetic diversity and geographic distance (r 2 = 0.014) on a small scale within the Chiloé Archipelago, as well as across the entire distribution of both species.
Table VI. Results of analysis of molecular variance (AMOVA) for each species of Crepipatella
Discussion
Analyses of DNA sequence data within and between species have been a powerful tool not only for determining aspects of the evolutionary history of the species, but also in illuminating the forces acting on life history traits. In this study, we found higher genetic diversity in C. dilatata than in C. fecunda (, ). Although there were only two species sampled, our results are consistent with other studies that have compared genetic diversity in invertebrate species with different developmental modes (Collin Citation2001; Zickovich & Bohonak Citation2007).
Comparisons of mitochondrial COI sequences also show that C. fecunda lacks genetic structure throughout most of the groupings and sampling locations (, ), in contrast to the significant structure exhibited by C. dilatata (, ). In general, lack of significant genetic structure can be attributed to high rates of gene flow among populations or recent colonization (Palumbi & Sotka Citation2006). In the context of the developmental mode, it is possible that the nature of the current patterns in the southeastern Pacific affect the migration of both species in different ways, and that these differences could be attributed to the contrasting dispersal capabilities of these two species. That is to say, the lack of larval dispersal in C. dilatata would allow local retention mechanisms to largely isolate this species allowing time for genetic differentiation. In fact, a significant proportion (56.86%) of the genetic variation in C. dilatata can be explained by the variance between populations ().
Although pooling populations for species with strong differentiation such as C. dilatata is expected to lead to an excess of medium-frequency alleles because of fixed differences among populations, for weakly differentiated species (such as C. fecunda), pooling generates an excess of low-frequency variants as singletons present within-population often remain singletons within the pool (Arunyawat et al. Citation2007). Based on our results, we suggest that the effect of population structure, in interaction with the sampling scheme (e.g Coquimbo with seven specimens), debilitate the population stasis/neutrality hypothesis based on pooled samples of C. dilatata. Further, the absence of fixed nucleotide differences between populations of C. fecunda implies that pooling the population samples does not result in additional internal branches of the genealogy, but yields an excess of singletons as most of the singletons within local populations still remain singletons in the pooled sample. This may partially explain the higher negative values observed for Tajima's D in the pooled sample of C. fecunda.
Consistent with the AMOVA analyses, results of phylogenetic analyses (), haplotype network () and mismatch distribution () indicate that the predominant demographic and phylogeographic patterns of C. fecunda are a recent population expansion and a lack of genetic structure throughout its distributional range of approximately 5000 km (Ancón, Perú to Melinka, Chile). Furthermore, we did not detect genetic differentiation at any spatial scale. The demographic and phylogeographic patterns of C. dilatata correspond to a multimodal mismatch distribution () consistent with long-term stationary populations (Rogers & Harpending Citation1992). The structure depicted by the haplotype network of this species () shows well-defined groups coincident with sampling locations (), with a reduced haplotypic and nucleotidic intrapopulational diversity, except for Puerto Madryn (). In spite of the expected association between haplotype and geographical location, there were haplotypes shared between Chile and Argentina, and the IBD test was not significant between both groupings. The lack of IBD signature between both geographical locations could be indicative of population connectivity favored by both the Humboldt counter current and West Drift current.
C. dilatata and C. fecunda occur in ecologically similar environments, and are sympatric at some locations (Gallardo Citation1979). If we accept that the presence of planktonic larvae is positively correlated with a wider dispersal and gene flow, one should find wider geographic ranges in species with planktonic larvae compared to those without them, but this is not the case when comparing C. dilatata and C. fecunda. C. dilatata has a wider geographic distribution than C. fecunda, occurring on southeastern Pacific and southwestern Atlantic coasts, while C. fecunda occurs only on southeastern Pacific coasts.
shows the resulting phylogenetic tree derived from the COI gene sequences. In both analyses (NJ and Bayesian) C. dilatata is subdivided into two groups and C. fecunda appears nested within the paraphyletic clade of C. dilatata. Same results are obtained in a molecular and embriological study reported by Collin et al. (Citation2007). The authors use parsimony, maximum likelihood and Bayesian tree reconstructions, and point out that the planktotrophy of C. fecunda has re-evolved from a direct developing ancestor. In other words, they suggest that the nesting of C. fecunda within the paraphyletic C. dilatata is indicative that the latter could be the extant ancestor of C. fecunda. This recent derivation of C. fecunda from C. dilatata is consistent with the distributional range and the population genetic patterns obtained in this study. It is further supported by independent genetic studies, which indicate a low genetic divergence between both species (Collin Citation2002, Citation2003; Véliz et al. Citation2003).
In marine environments, genetic connectivity has been mainly related to the mode of larval development and biogeographic barriers (Teske et al. Citation2008) in which brooding or viviparous species (i.e. with no pelagic larval dispersal) tend to be highly subdivided, suggesting very low gene flow among these populations. That generalization also suggests that species with larvae that spend longer in the pelagic environment will have greater gene flow, greater dispersal capabilities, lower genetic differentiation and better-connected populations (Hellberg Citation2009). However, exceptions have been documented, such as the copepod Tigriopus californicus (Burton & Feldman Citation1981), the tropical corals Goniastrea favulus and Platygyra daedalea (Miller & Ayre Citation2008), and gastropods of the genus Littorina (Kyle & Boulding Citation2000), among others. In this line, Johannesson (Citation1998) suggests that the passive transport of adult benthic invertebrates of low mobility in combination with a direct development may be a more powerful means of long-distance dispersal than larval spreading, in at least certain cases. This implies that direct developers, which are poor dispersers on short distances may be better off than planktonic developers on long distances.
Finally, we would like to point out that the results of this study are based solely on a single gene and depict the history of the mitochondrial COI lineage, but not necessarily that of the species. For example, recent shifts in populations due to glacial cycles like the South American Last Maximum Glacial (Latorre et al. Citation2007) may have obscured genetic differentiation and give the impression of high contemporary larval exchange in C. fecunda. Moreover, the networks themselves may not reflect the true genealogical history, due to homoplasious mutations that introduce uncertainty in the reconstruction. Thus, the probability of specific scenarios resulting in the observed data could be unquantified. For example, further analyses are necessary to evaluate the possibility that the shared haplotypes between the Coquimbo and Puerto Madryn populations of C. dilatata could be homoplasy, lineage sorting or ancestral polymorphisms. No doubt, integrating our developmental and population genetics results with independent ecological and phylogenetic studies would strengthen the inferences concerning the evolutionary processes that underlie the population genetic patterns of C. dilatata and C. fecunda.
Acknowledgements
We are grateful to Dr. Oscar Chaparro (Instituto de Biología Marina, UACH) for providing us with some samples from Quempillén and Patricia Vallejos for samples from Aucho. We also appreciate the comments of Dr. Carlos Jara that improved the manuscript. This study was partially supported by DID-UACh S 2005–13 to JJN, additionally by DIAT UMAG-0273 and DID UACH S 2007–57 research projects to CGS.
References
- Arndt , A and Smith , J. 1998 . Genetic diversity and population structure in two species of sea cucumber: Differing patterns according to mode of development . Molecular Ecology , 7 : 1053 – 1064 .
- Arunyawat , U , Stephan , W and Stadler , T. 2007 . Using multilocus sequence data to assess population structure, natural selection, and linkage disequilibrium in wild tomatoes . Molecular Biology and Evolution , 24 : 2310 – 2322 .
- Ayre , DJ and Hughes , TP. 2000 . Genotypic diversity and gene flow in brooding and spawning corals along the Great Barrier Reef, Australia . Evolution , 54 : 1590 – 1605 .
- Bandelt , HJ , Forster , P and Röhl , A. 1999 . Median-joining networks for inferring intraspecific phylogenies . Molecular Biology and Evolution , 16 : 37 – 48 .
- Burton , RS and Feldman , MW. 1981 . Population genetics of Tigriopus californicus. II. Differentiation among neighboring populations . Evolution , 35 : 1192 – 1205 .
- Chaparro , OR , Charpentier , JL and Collin , R. 2002 . Embryonic velar structure and function of two sibling species of Crepidula with different modes of development . Biological Bulletin , 203 : 80 – 86 .
- Chaparro , OR , Saldivia , CL , Pereda , SV , Segura , CJ , Montiel , YA and Collin , R. 2005 . The reproductive cycle and development of Crepipatella fecunda (Gastropoda: Calyptraeidae) from southern Chile . Journal of the Marine Biological Association of the United Kingdom , 85 : 157 – 161 .
- Collin , R. 2001 . The effects of mode of development on phylogeography and population structure of North Atlantic Crepidula (Gastropoda: Calyptraeidae) . Molecular Ecology , 10 : 2249 – 2262 .
- Collin , R. 2002 . Another last word on Crepidula convexa and a description of C. ustulatulina sp. nov. (Gastropoda: Calyptraeidae) from the Gulf of Mexico . Bulletin of Marine Science , 70 : 177 – 184 .
- Collin , R. 2003 . Phylogenetic relationships among calyptraeid gastropods and their implications for the biogeography of speciation . Systematic Biology , 52 : 618 – 640 .
- Collin , R , Chaparro , O , Winkler , F and Véliz , D. 2007 . Molecular phylogenetic and embryological evidence that feeding larvae have been reacquired in a marine gastropod . Biological Bulletin , 212 : 83 – 92 .
- Duffy , JE. 1993 . Genetic population structure in two tropical sponge-dwelling shrimps that differ in dispersal potential . Marine Biology , 90 : 127 – 138 .
- Dupanloup , I , Schneider , S and Excoffier , L. 2002 . A simulated annealing approach to define the genetic structure of populations . Molecular Ecology , 11 : 2571 – 2581 .
- Excoffier , L. 2004 . Patterns of DNA sequence diversity and genetic structure after a range expansion: Lessons from the infinite-island model . Molecular Ecology , 13 : 853 – 864 .
- Excoffier , L , Smouse , PE and Quattro , JM. 1992 . Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data . Genetics , 13 : 479 – 491 .
- Felsenstein , J. 1985 . Confidence limits on phylogenies: An approach using the bootstrap . Evolution , 39 : 783 – 791 .
- Folmer , O , Black , M , Hoeh , W , Lutz , R and Vrijemhoek , R. 1994 . DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates . Molecular Marine Biology and Biotechnology , 3 : 294 – 299 .
- Fu , YX and Li , WH. 1993 . Statistical tests of neutrality of mutations . Genetics , 133 : 693 – 709 .
- Gallardo , CS. 1977 . Two modes of development in the morphospecies Crepidula dilatata (Gastropoda: Calyptraeidae) from Southern Chile . Marine Biology , 39 : 241 – 251 .
- Gallardo , CS. 1979 . Especies gemelas del género Crepidula (Gastropoda, Calyptraeidae) en la costa de Chile; una redescripción de C. dilatata Lamarck y descripción de C. fecunda n. sp . Studies on the Neotropical Fauna and Environment , 14 : 215 – 226 .
- Harpending , HC. 1994 . Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution . Human Biology , 66 : 591 – 600 .
- Hellberg , ME. 1996 . Dependence of gene flow on geographic distance in two solitary corals with different larval dispersal capabilities . Evolution , 28 : 1167 – 1175 .
- Hellberg , ME. 2009 . Gene flow and isolation among populations of marine animals . Annual Review of Ecology Evolution and Systematics , 40 : 291 – 310 .
- Huelsenbeck , JP and Ronquist , F. 2003 . MrBayes 3: Bayesian phylogenetic inference under mixed models . Bioinformatics , 19 : 1572 – 1574 .
- Iuri , V , Patti , FP and Procaccini , G. 2007 . Phylogeography of the sea urchin Paracentrotus lividus (Lamarck) (Echinodermata: Echinoidea): First insights from the South Tyrrhenian Sea . Hydrobiologia , 580 : 77 – 84 .
- Janson , K. 1987 . Allozyme and shell variation in two marine snails (Littorina, Prosobranchia) with different dispersal abilities . Biological Journal of the Linnean Society , 30 : 245 – 256 .
- Jensen JL, Bohonak AJ, Kelley ST. 2005. Isolation by distance, web service. BMC Genetics 6:13 http://ibdws.sdsu.edu/ (http://ibdws.sdsu.edu/)
- Johannesson , K. 1998 . The paradox of Rockall: Why is a brooding gastropod (Littorina saxatilis) more widespread than one having planktonic larval dispersal stage (L. littorea)? . Marine Biology , 99 : 507 – 513 .
- Katoh , K , Kuma , K , Toh , H and Miyata , T. 2005 . MAFFT version 5: Improvement in accuracy of multiple sequence alignment . Nucleic Acids Research , 33 : 511 – 518 .
- Kelly RP , Palumbi SR. 2010 . Genetic structure among 50 species of the Northeastern Pacific rocky intertidal community . PLoS ONE 5 : e8594 . doi:10.1371 .
- Kyle , C and Boulding , EG. 2000 . Comparative population genetic structure of marine gastropods (Littorina spp.) with and without pelagic larval dispersal . Marine Biology , 137 : 835 – 845 .
- Latorre , C , Moreno , PI , Vargas , G , Maldonado , A , Villa-Martínez , R Armesto , JJ . 2007 . “ Late Quaternary environments and palaeoclimate ” . In Geology of Chile , Edited by: Gibbons , W and Moreno , T . 309 – 328 . London : Geological Society Press . Ineditorspp
- Levin , LA. 2006 . Recent progress in understanding larval dispersal: New directions and digressions . Integrative and Comparative Biology , 46 : 282 – 297 .
- Librado , P and Rozas , J. 2009 . DnaSP v5: A software for comprehensive analysis of DNA polymorphism data . Bioinformatics , 25 : 1451 – 1452 .
- Miller , KJ and Ayre , DJ. 2008 . Population structure is not a simple function of reproductive mode and larval type: Insights from tropical corals . Journal of Animal Ecology , 7 : 713 – 724 .
- Nei , M. 1987 . Molecular evolutionary genetics , 512 New York, NY : Columbia University Press .
- Palumbi , SR and Sotka , EE. 2006 . The use of genetic clines to estimate dispersal distances of marine larvae . Ecology , 87 : 1094 – 1103 .
- Peterson , MA and Denno , RF. 1998 . The influence of dispersal and diet breadth on patterns of genetic isolation by distance in phytophagous insects . American Naturalist , 152 : 428 – 446 .
- Ramos-Onsins , SE and Rozas , J. 2002 . Statistical properties of new neutrality tests against population growth . Molecular Biology and Evolution , 19 : 2092 – 2100 .
- Rogers , AR and Harpending , H. 1992 . Population growth makes waves in the distribution of pairwise genetic differences . Molecular Biology and Evolution , 9 : 552 – 569 .
- Sambrook , J , Fritsch , EF and Maniatis , T. 1989 . Molecular cloning: A laboratory manual , 2nd , New York, NY : Cold Spring Harbour Laboratory .
- Schneider , S , Kueffer , JM , Roessli , D and Excoffier , L. 2006 . “ Arlequin: A software for population genetic data analysis ” . In Genetics and Biometry Laboratory , Switzerland : University of Geneva .
- Tajima , F. 1989 . Statistical method for testing the neutral mutation hypothesis by DNA polymorphism . Genetics , 123 : 585 – 595 .
- Teske , PR , Papadopoulos , I , Newman , BK , Dworschak , PC , McQuaid , CD and Barker , NP. 2008 . Oceanic dispersal barriers, adaptation and larval retention: An interdisciplinary assessment of potential factors maintaining a phylogeographic break between sister lineages of an African prawn . BMC Evolutionary Biology , 8 : 341
- Véliz , D , Winkler , F and Guisado , C. 2003 . Developmental and genetic evidence for the existence of three morphologically cryptic species of Crepidula in northern Chile . Marine Biology , 143 : 131 – 142 .
- Watts , PC and Thorpe , JP. 2006 . Influence of contrasting larval developmental types upon the population-genetic structure of cheilostome bryozoans . Marine Biology , 149 : 1093 – 1101 .
- Wilke , T and Davis , GM. 2000 . Infraspecific mitochondrial sequence diversity in Hydrobia ulvae and Hydrobia ventrosa (Hydrobiidae: Rissooidea: Gastropoda): Do their different life histories affect biogeographic patterns and gene flow? . Biological Journal of the Linnean Society , 70 : 89 – 105 .
- Xia , X and Xie , Z. 2001 . DAMBE: Data analysis in molecular biology and evolution . Journal of Heredity , 92 : 371 – 373 .
- Xia , X , Xie , Z , Salemi , M , Chen , L and Wang , Y. 2003 . An index of substitution saturation and its application . Molecular Phylogenetics and Evolution , 26 : 1 – 7 .
- Zickovich , J and Bohonak , AJ. 2007 . Dispersal ability and genetic structure in aquatic invertebrates: A comparative study in southern California streams and reservoirs . Freshwater Biology , 52 : 1982 – 1996 .