
Abstract
The monokaryotic strain, Schizophyllum commune strain IUM1114-SS01, was generated from a basidiospore of dikaryotic parental strain IUM1114. It even showed the decolorizing activities for several textile dyes much better than its parental strain. Based on the results of a single-molecule real-time sequencing technology, we present the draft genome of S. commune IUM1114-SS01, comprising 41.1 Mb with GC contents of the genome were 57.44%. Among 13,380 protein-coding genes, 534 genes are carbon hydrate-active enzyme coding genes.
The basidiomycetes fungus Schizophyllum commune has been used as a model of mushroom development because it takes short time for fruiting body formation on synthetic media, its whole genome sequence is publicly available, and many molecular tools are applicable [Citation1]. In addition, this fungus is easily found on trees where it decomposes recalcitrant lignin which has similar structure appear in some of hard to degrade synthetic dyes [Citation2,Citation3]. Therefore, studying the dye degradation ability on S. commune species is a promising area, to be manipulated for using in industrial process such as biodegradation of complex substances or pollutants, processing agricultural products or byproducts of biofuel production in the future.
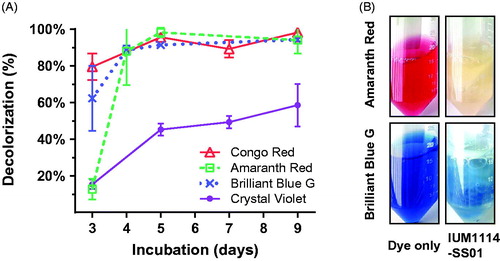
Recently, we screened 81 S. commune strains deposited in the Culture Collection of Mushrooms in Incheon National University for the decolorizing activities for several textile dyes and found one excellent candidate with decolorization for all dyes named as IUM1114 [Citation4]. From this strain, total 64 single basidiospore were isolated and among them, 20 single spores were subjected for testing of mating ability. Within 400 combinations, there were 21 combinations showing the success of mating. The 20 strains were also grown in aerated media using filtered vent cap falcon tubes supplied with different textile dyes, such as: Congo Red, Crystal Violet, Amaranth Red, Brilliant Blue G. One of the excellent single strains that were highly responsive to dye-decolorization were IUM1114-SS01 (). This strain expressed the dye decolorization ability from 50% up to 97% with the dyes: Crystal Violet, Amaranth Red, Brilliant Blue G and Congo Red (). The red color of Amaranth Red dye almost disappearing after 10 days of incubation with fungus suggested the decolorization ability of this strain on Amaranth Red. While the blue color of Brilliant Blue G dye just remained in fungus clumps but not in the solution implied the dye absorption ability of this strain (). The whole-genome de novo sequencing of this S. commune IUM1114-SS01 was performed to further understand the dye-decolorizing enzyme systems of this fungus.
Figure 1. Decolorization ability of S. commune IUM1114-SS01 in aerated liquid PDB. (A) The decolorization ability of each sample was measured from 3 days after inoculation with dye in filtered vent cap falcon tube. (B) Pictures of color changes in Brilliant Blue G and Amaranth Red solutions were taken after 10 days of inoculation. Control sample is the PDB solution supplemented with equivalent dye.
Genomic DNA of S. commune IUM1114-SS01 was extracted from homogenized mycelia of the 3-day-old subculture on potato dextrose agar at 25 °C using Nucleospin® Plant II kit from Macherey-Nagel company according to manufacturer’s instruction. Genome sequencing was performed using PacBio single-molecule real-time (SMRT) sequencing technology. High-molecular-weight genomic DNA was sheared and selected on a BluePippin system using a cutoff range of >15 kb. The libraries were sequenced on one SMRT cells using a PacBio Sequel platform according to the manufacturer’s instructions (Pacific Biosciences, Menlo Park, CA, USA). For de novo assembly, a total of 7.2 billion bases from 753,103 reads were assembled using the SMRT Analysis Hierarchical Genome Assembly Process 3 (HGAP3) pipeline, which resulted in 84 contigs, with the length of the N50 value being 19,27,764 bp. In addition, a total of 4.9 billion bases from 32.5 million reads were sequenced for error correction through the Illumina HiSeq 4000 platform. The HiSeq reads were applied for accurate genome sequence by using Pilon (v1.21), resulting in 74 contigs with the length of the longest one and the N50 value being 44,38,218 bp and 19,27,765 bp, respectively (). The total size of the S. commune IUM1114-SS01 genome was 4,10,06,423 bp and the cumulative GC content of the genome was 57.44% ().
Table 1. Summary of the genome assembly and annotation of S. commune IUM1114-SS01.
Ab initio gene prediction was carried out using Maker v2.31.10 [Citation5] based on gene model training from S. commune reference strain (H4-8). As a result, 13,386 protein encoding genes were predicted including 214 nuclear tRNAs (). Genes were functionally characterized using BLAST+ (v2.7.1+) [Citation6] and InterProScan [Citation7]. The carbohydrate-active enzyme (CAZyme) gene content was determined using dbCAN2 v2.0.6 [Citation8, Citation9] based on three different tools, HMMER v3.2.1, DIAMOND v0.9.30, and Hotpep [Citation10]. The analysis revealed that the S. commune IUM1114-SS01 genome contains 249 glycoside hydrolases (GH), 91 glycosyltransferases (GT), 18 polysaccharide lyases (PL), 66 carbohydrate esterases (CE), 18 carbohydrate-binding modules (CBM), and 89 enzymes with auxiliary activities (AA) which are similar with S. commune H4-8 strain () [Citation1]. Move over, the genome sequence of this strain has the highest number of total CAZymes and glycoside hydrolases (GH) compared to the genomes of other closely related Agaricales species () [Citation11–14]. These indicated that this S. commune IUM1114-SS01 strain has high potential to be used for biomass conversion.
Table 2. Comparison of the number of CAZymes of S. commune IUM1114-SS01 with those of other fungi in the order Agaricales.
The genomic information of S. commune IUM1114-SS01 will provide a better understanding of the genomic network involved in dye degrading metabolic process in the white rot fungi. The draft genome sequence of S. commune IUM1114-SS01 has been deposited in GenBank under the accession number JAATOI000000000, BioProject number PRJNA615298, and BioSample number SAMN14450467.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Ohm RA, de Jong JF, Lugones LG, et al. Genome sequence of the model mushroom Schizophyllum commune. Nat Biotechnol. 2010; 28(9):957–963.
- Hatakka A. Lignin-modifying enzymes from selected white-rot fungi: production and role from in lignin degradation. FEMS Microbiol Rev. 1994;13(2–3):125–135.
- Dwivedi P, Tomar RS. Bioremediation of textile effluent for degradation and decolourization of synthetic dyes: a review. Int J Curr Res Life Sci. 2018;7(04):1948–1951.
- Choi Y, Nguyen Y, Lee TS, et al. Genetic diversity and dye-decolorizing spectrum of Schizophyllum commune population. J Microbiol Biotechnol. 2020;30(10):1525–1535.
- Cantarel BL, Korf I, Robb SMC, et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18(1):188–196.
- Camacho C, Coulouris G, Avagyan V, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10(1):421.
- Jones P, Binns D, Chang H-Y, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30(9):1236–1240.
- Zhang H, Yohe T, Huang L, et al. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018;46(W1):W95–W101.
- Yin Y, Mao X, Yang J, et al. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40(Web Server issue):W445–W451.
- Busk PK, Pilgaard B, Lezyk MJ, et al. Homology to peptide pattern for annotation of carbohydrate-active enzymes and prediction of function. BMC Bioinformatics. 2017;18(1):214.
- Morin E, Kohler A, Baker AR, et al. Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc Natl Acad Sci U S A. 2012;109(43):17501–17506.
- Stajich JE, Wilke SK, Ahrén D, et al. Insights into evolution of multicellular fungi from the assembled chromosomes of the mushroom Coprinopsis cinerea (Coprinus cinereus). Proc Natl Acad Sci U S A. 2010;107(26):11889–11894.
- Martin F, Aerts A, Ahrén D, et al. The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature. 2008;452(7183):88–92.
- Riley R, Salamov AA, Brown DW, et al. Extensive sampling of basidiomycete genomes demonstrates inadequacy of the white-rot/brown-rot paradigm for wood decay fungi. Proc Natl Acad Sci U S A. 2014;111(27):9923–9928.