
Abstract
The coastal marine ecosystems of Vietnam are one of the global biodiversity hotspots, but the biodiversity of marine fungi is not well known. To fill this major gap of knowledge, we assessed the genetic diversity (ITS sequence) of 75 fungal strains isolated from 11 surface coastal marine and deeper waters in Nha Trang Bay and Van Phong Bay using a culture-dependent approach and 5 OTUs (Operational Taxonomic Units) of fungi in three representative sampling sites using next-generation sequencing. The results from both approaches shared similar fungal taxonomy to the most abundant phylum (Ascomycota), genera (Candida and Aspergillus) and species (Candida blankii) but were different at less common taxa. Culturable fungal strains in this study belong to 3 phyla, 5 subdivisions, 7 classes, 12 orders, 17 families, 22 genera and at least 40 species, of which 29 species have been identified and several species are likely novel. Among identified species, 12 and 28 are new records in global and Vietnamese marine areas, respectively. The analysis of enzyme activity and the checklist of trophic mode and guild assignment provided valuable additional biological information and suggested the ecological function of planktonic fungi in the marine food web. This is the largest dataset of marine fungal biodiversity on morphology, phylogeny and enzyme activity in the tropical coastal ecosystems of Vietnam and Southeast Asia. Biogeographic aspects, ecological factors and human impact may structure mycoplankton communities in such aquatic habitats.
1. Introduction
Vietnamese coastal marine ecosystem is one of the most biologically diverse marine ecosystems, overlapping with the Coral Triangle and 4 out of 35 global biodiversity hotspots with unique biota. This region is identified as a high-priority area for marine conservation [Citation1]. However, the marine ecosystems of Vietnam are severely impacted by climate change [Citation2] and anthropogenic pollutions [Citation3–7]. Severe ecological impacts from anthropogenic disturbance on the marine biodiversity of Vietnam have been observed in recent years [Citation3,Citation7,Citation8]. Among marine organisms, fungi play a critical role in biogeochemical cycles [Citation9,Citation10]. Assessing the biodiversity of marine fungi in the coastal environment of Vietnam is the first but critical step to provide a scientific base for conservation as rapid biodiversity loss is observed in this region [Citation11]. Furthermore, it opens the opportunity to explore bioactive compounds from marine fungi that may have great pharmacological and industrial applications [Citation12–16]. However, the biodiversity of marine fungi and their ecological roles in the coastal environment of Vietnam is largely unknown.
A marine fungus is defined as “any fungus that is recovered repeatedly from marine habitats and: 1) is able to grow and/or sporulate (on a substrate) in marine environments; 2) forms symbiotic relationships with other marine organisms; or 3) is shown to adapt and evolve at the genetic level or be metabolically active in marine environments” [Citation17]. The updated classification of marine fungi has 1,112 species, 472 genera, 129 families and 65 orders. Major phyla include Ascomycota, Basidiomycota, Chytridiomycota (chytrids) and related phyla, Zygomycota, and Blastocladiomycota. The Halosphaeriaceae remains the largest family of marine fungi with 141 species in 59 genera, while the most specious genera are Candida (64 species), Aspergillus (47 species), and Penicillium (39 species) [Citation18]. The members of Zygomycota are now part of two phyla Mucoromycota and Zoopagomycota, and mostly terrestrial in habitat [Citation19]. Marine fungi were retrieved exclusively from the sea surface to the depth of kilometers beneath the ocean sediments. Despite their diverse roles in nutrition cycles and food webs, as well as their commensal interaction or pathogenicity to other organisms, little is known about the diversity of this major eukaryotic branch in their marine ecosystems or their ecological functions [Citation9].
The diversity of marine plankton fungi has been studied from a few coastal locations such as Portugal [Citation20], Taiwan [Citation21], India [Citation22], Chile [Citation23,Citation40], Brazil [Citation24], France [Citation25], and China [Citation26,Citation27]. A few notable studies were performed, e.g., molecular analysis across 130 European marine samples [Citation28]. More studies were reported on fungi in the marine environments associated with animals, plants and algae [Citation29–36] and in deepsea waters [Citation37–42]. These molecular studies have retrieved marine fungal sequences belonging to Ascomycota, Basidiomycota, and Chytridiomycota, and some of them were grouped into novel environmental clusters.
In this study, we comprehensively assessed the biodiversity of fungi in the coastal marine habitats in Nha Trang Bay and Van Phong Bay of the Central Vietnam region where is experiencing a rapid increase in anthropogenic disturbances from urbanization, tourism, shipping, and aquaculture development that may seriously affect the marine environment and accelerate biodiversity loss [Citation6,Citation11,Citation43–46]. Importantly we used two different methods that are culture-dependent and culture-independent approaches that may provide complementary results to have a more comprehensive assessment of marine fungal biodiversity. Culturable fungal strains were identified based on ITS sequence, followed by the description of morphology, phylogeny and taxonomy of major fungal classes and orders. To provide valuable additional biology information and mechanistically assess the ecological role of fungi in the marine ecosystems, we analyzed the total relative enzyme activity and checklist of trophic mode and guild assignment.
2. Materials & methods
2.1. Site description and sampling strategy
A total of 11 seawater samples were collected from coastal marine habitats of Nha Trang Bay and Van Phong Bay at Khanh Hoa Province, Vietnam from May to August 2017 (, ) using a 2.2-liter sterile sampler (Horizontal Beta Bottle style, Wildco, Yulee, Florida, USA). Water samples were carried in 1.5 liters sterile, screw-capped plastic bottles, stored in ice and transferred immediately to the laboratory after collection. Also, the temperature, pH, and salinity of the water samples were monitored at each sampling site using a multiparameter meter. The sample sets were randomly split into two subsamples to employ the two culture-dependent and culture-independent approaches simultaneously.
Figure 1. Sampling positions at Nha Trang Bay and Van Phong Bay of Khanh Hoa Province, Vietnam. Detail information on locations was given in the .
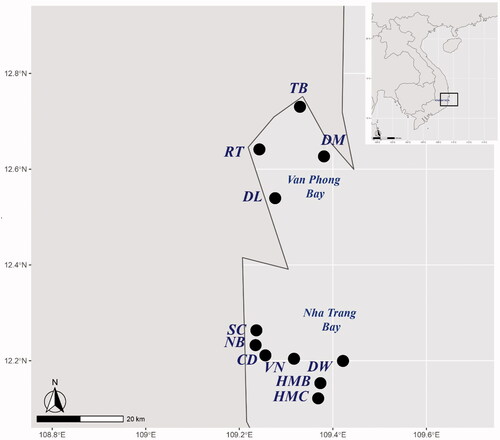
Table 1. Details regarding colony forming units (CFUs) of fungal colonies on the SDA medium plates and strains identified with ITS sequence.
2.2. Culture-dependent approach
2.2.1. Isolation and identification of marine fungi
Fungi from the seawater samples were isolated within three hours of collection using the membrane filtration technique [Citation26]. Briefly, seawater samples were diluted into concentrations at 10−1, 10−2, and 10−3. Subsequently, diluted water samples (15 ml, triplicate) were filtered through sterile 0.45 µm cellulose esters membrane (MilliporeSigma, Burlington, Massachusetts, USA). These membranes were then placed on solid media plates of Sabouraud Dextrose Agar (SDA) (Thermo Fisher Scientific, Waltham, Massachusetts, USA) supplemented with antibiotics (0.075% streptomycin sulfate and 0.05% ampicillin) (Thermo Fisher Scientific) to suppress bacterial growth. The plates were incubated at 28 °C for 2–3 days and examined daily for the growth of fungi. Fungal colonies that developed were subcultured onto fresh Potato Dextrose Agar (PDA) (Thermo Fisher Scientific) plate (for filamentous fungi) or Yeast Extract Peptone and Dextrose (YPD) (Thermo Fisher Scientific) plate (for yeast) for pure, single colony isolation and identification. Filamentous fungi were identified based on macroscopic and microscopic morphological characteristics (Figure S1, Supplementary Material). Strains were named according to the name of the sampling location, the order of strains isolated from each site, and a final symbol distinguishing between yeast (Y) and mold (M) (Table S1, Supplementary Material). For example, the strain VN1Y means that it was sampled at Vung Ngan (VN) as the first (1) strain isolated from this site and it is a yeast (Y). Fungal strains with different morphology have been selected for further analysis and deposited in Microbiological Culture Collection at Nha Trang University (NTU), Vietnam with isolate IDs shown in Table S1 (Supplementary Material).
2.2.2. DNA isolation, PCR and sequencing
For DNA isolation, yeasts were grown in a YPD broth medium and shaken at 170 rpm for 1–3 days at 25–28 °C, whereas a PDA plate was used for growing filamentous fungi for 3–5 days at the same temperature. Yeast cells were harvested by centrifugation and genomic DNA was extracted according to the method of Harju et al. [Citation47]. For mold, mycelia were harvested, lyophilized and crushed in a mortar with pestle using liquid nitrogen to a fine powder. Genomic DNA from mold isolates was extracted using the EZ − 10 Spin Column Plant Genomic DNA Miniprep Kit (Bio Basic, Canada) as directed by the manufacturer. The extracted DNA samples were stored at −20 °C.
The ITS domain of nuclear ribosomal genes was amplified by polymerase chain reaction (PCR) using the appropriate specific primers to target Ascomycota and Basidiomycota (ITS1-F_KYO2: 5′-TAG AGG AAG TAA AAG TCG TAA-3′ [Citation48] and ITS4-R: 5′-TCC TCC GCT TAT TGA TAT GC-3′ [Citation49]). The PCR method was performed as described previously [Citation46,Citation50] with some modifications. Briefly, one microliter of DNA (∼25 ng) was added to 50 µl reaction volume containing 1 µl of Taq polymerase (Bioline, Memphis, Tennessee, USA), 10 µl of 5X MyTaq reaction buffer, 35 µl of distilled deionized water and 10 pmoles of each primer. The PCR program of ITS rRNA gene was run for initial denaturation step at 93 °C for 3 min, followed by 35 cycles of 0.5 min at 93 °C, 0.5 min at 55-60 °C and 0.5 min at 72 °C, and a final extension at 72 °C for 5 min. The amplified products were separated on a 1% (w v−1) agarose gel stained with ethidium bromide and visualized under a UV transilluminator.
PCR products were purified using the QIAquick PCR purification Kit (Qiagen, Hilden, Germany), and then Sanger sequenced at Macrogen (Soul, Korea) in both directions with the same PCR primers as described above using Big Dye terminator in a 3730 × l DNA Analyzer (Applied Biosystems, Waltham, Massachusetts, USA). The DNA sequences generated in this study are deposited in GenBank under the accession numbers from MT102787 to MT102861 for the ITS gene.
2.2.3. Sequence alignment and phylogenetic analyses
Partial gene sequences of fungi samples obtained in this study and reference sequences available in GenBank were used for sequence analysis at the National Center for Biotechnology Information (NCBI) using BLASTn (http://www.ncbi.nlm.nih.gov/BLAST). Gene sequences were aligned using MUSCLE [Citation51], and regions with gaps were removed using BioEdit [Citation52]. Model selection was used to determine the best fit model with the lowest Bayesian Information Criterion score for Maximum Likelihood, Minimum Evolution, and Neighbor-Joining method [Citation53], which was then used to construct a phylogenetic tree using the MEGA X program [Citation54]. The evolutionary distances were computed by using the Kimura 2-parameter model and Tamura 3-parameter model [Citation55]. The robustness of the tree topology was tested by bootstrap analysis with 1000 re-samplings [Citation56].
2.3. Culture-independent approach
2.3.1. DNA extraction from seawater samples for metabarcoding
As the three seawater samples HMC, DW and CD showed the highest density of marine fungi by culture-dependent approach at Nha Trang Bay, they were selected as 3 representatives for culture-independent studies (). Seawater samples were filtered through sterile 0.45 µm cellulose ester membranes (MilliporeSigma, Burlington, Massachusetts, USA). DNA samples were extracted directly from filters using the PowerWater DNA Isolation Kit (Qiagen) according to the manufacturer’s instructions. All the DNA samples were quantitated and quality checked with the NanoDrop ND-2000C spectrophotometer (ThermoFisher Scientific, Dreieich, Germany). DNA extracts were then stored at −20 °C for further analysis.
2.3.2. Marine fungi analysis: Sequencing
Metagenome amplicon libraries were generated from DNA extracts and sequenced at Macrogen (Soul, Korea) using the standard protocol as described in the manufacturer’s instructions (Illumina, San Diego, California, USA). Briefly, amplification of ITS regions was performed using the sequence-specific primers (ITS1-F_KYO2 and ITS4-R) described above in a two-step dual-indexed PCR approach designed specifically for Illumina instruments, in which the gene-specific sequence was fused to the Illumina sequencing primers. The quantity of DNA was checked by picogreen method with Quant-iT™ PicoGreen™ dsDNA Assay Kit (Introgene, Leiden, Netherlands) using Victor 3 fluorometry. The library quality was assessed on an Agilent Technologies 2100 Bioanalyzer using a DNA 1000 chip to verify the size of PCR enriched fragments. For library quantity check, qPCR was used according to the Illumina qPCR Quantification Protocol Guide to achieving the highest quality of data on Illumina sequencing platforms. To generate a standard curve of fluorescence readings and calculate the library sample concentration, Roche's Rapid library standard Quantification solution and calculator were used. At last, the library was sequenced on an Illumina MiSeq2500 platform and 301 bp paired-end reads were generated.
2.3.3. Marine fungi analysis: Bioinformatic analysis
Paired-end reads were assigned to samples based on their unique barcode and truncated by cutting off the barcode and primer sequence. Paired-end reads were merged using FLASH (V1.2.11, http://ccb.jhu.edu/software/FLASH/) [Citation57] to significantly improve read assemblies.
To remove noise data and generate OTU clusters, assembled reads were pre-processed and clustered using CD-HIT-OTU (http://weizhongli-lab.org/cd-hit-otu/) [Citation58] according to the User’s Guide (http://weizhong-lab.ucsd.edu/cd-hit-otu/wiki/doku.php?id=cd-hit-otu_user_guide). Briefly, a three-step clustering was used for identifying OTUs. The first-step clustering was raw read filtering and trimming. The second step was error-free reads picking. In the last step, OTU clustering was generated at different distance cutoffs (0.03).
The program QIIME [Citation59] with script assign_taxonomy.py (http://qiime.org/scripts/assign_taxonomy.html) was used to assign taxonomy of the representative OTUs against the Unite Database (https://unite.ut.ee) [Citation60] using the Blast algorithm (≥80% identity and coverage).
The ITS fragments were extracted from both the Sanger sequencing (traditional) and Illumina sequencing datasets and the sequence similarity-based comparison was performed using the direct matching based on the Blast algorithm (≥95% identity and coverage) at the species level.
The eukaryote ITS rDNA genes Illumina sequencing data are deposited in the NCBI under the BioProject with GenBank accession numbers from MT364507 to MT364634.
2.4. Screening for extracellular enzyme activities
The fungal isolates were screened using a qualitative plate assay for different enzymes (protease, amylase, phytase, cellulase and chitinase) by streaking them on the modified Czapek-Dox agar plates, supplemented with ampicillin (0.05%) and streptomycin sulfate (0.075%) as well as specific substrates. Modified Czapek-Dox agar medium included NaNO3 2 g L−1, K2HPO4 1 g L−1, MgSO4 7H2O 0.5 g L−1, KCl 0.5 g L−1, FeSO4 7H2O 0.01 g L−1, agar-agar 15 g L−1, at pH 6.5 ± 0.2. Sterile seawater was used at 50% (w w−1).
Protease activity was detected using modified Czapek–Dox agar plates after substitution NaNO3 with 1.0% skim milk [Citation61]. The fungal isolates were grown on these media plates at 28 °C for 5 days. The diameter of the clear zone around the colonies indicated protease activity.
Amylase activity was detected using modified Czapek–Dox agar plates supplemented with 1.0% soluble starch as the sole carbon source [Citation62]. The fungal isolates were grown on these media plates at 28 °C for 5 days. Then all culture plates (duplicates) were flooded with Lugol’s iodine solution. The diameter of the clear zone surrounding the colonies indicated amylase activity.
Phytase activity was detected using modified Czapek-Dox agar plates supplemented with sodium phytate (Sigma-Aldrich, St. Louis, Missouri, USA) at the concentration of 10 g L−1 [Citation63]. The fungal isolates were grown on these media plates at 28 °C for 3 days. Then, petri dishes were flooded with Lugol’s iodine solution. The diameter of the clear zone around the colonies indicated phytase activity.
Cellulase activity was detected using modified Czapek-Dox agar plates supplemented with 0,5% cacboxylmethyl cellulose [Citation64]. The fungal isolates were grown on these media plates at 28 °C for 4 days. Then, petri dishes were flooded with an aqueous solution of 0.1% Congo red and shaken at 80 rpm for 15 min on a rotary shaker. The diameter of the clear zone around the colonies on media plates indicated cellulase activity.
Chitinase activity was detected using modified Czapek–Dox agar plates supplemented with colloidal chitin 10 g L−1 [Citation65]. The fungal isolates were grown on these media plates at 28 °C for 7 days. The diameter of the clear zone around the colonies on media plates indicated chitinase activity.
2.5. Fungal functional assignment and checklist
Fungal functional guilds were assigned using an open annotation tool (FUNGuild) [Citation66, online version: http://www.funguild.org/). Only the guild assignment with at least “possible” confidence rankings was accepted. Distribution and disease notes were confirmed and checklists from Fungal Databases of U.S. National Fungus Collections [Citation67] and previous studies published on National Center for Biotechnology Information (https://pubmed.ncbi.nlm.nih.gov/) by 23 March 2021.
2.6. Statistic analysis
The program QIIME [Citation59] with scripts such as alpha_diversity.py (http://qiime.org/scripts/alpha_diversity.html) was used to calculate alpha diversity (OTUs, Chao1, Shannon, Simpson) related to fungal OTU richness; alpha_rarefaction.py (http://qiime.org/scripts/alpha_rarefaction.html) to produce rarefaction curve graph; and make_2d_plots.py (http://qiime.org/scripts/make_2d_plots.html) to make 2 D PCoA Plots.
3. Results
3.1. Fungi density and molecular identification in the coastal ecosystems of Vietnam: Culture-dependent approach
The density of fungi of 11 surfaces coastal marine and deeper waters at Nha Trang Bay and Van Phong Bay ranged from 102 CFU L−1 to 105 CFU L−1 (). The lowest density of fungi was found in NB (6.0 × 102 CFU L−1) at Nha Trang Bay and DL (9.3 × 102 CFU L−1) at Van Phong Bay (). The Hon Mun Marine Protected Area of Nha Trang Bay had the highest fungal density, namely 1.8 × 105 CFU L−1 in the buffer zone and 1.1 × 105 CFU L−1 in the core zone HMC (), which were two orders of magnitude higher than the fungal density in NB and DL.
Overall, a total of 163 fungal colonies, including 115 yeasts and 48 filamentous fungi, were recovered on SDA medium in all samples with differences in depth, pH, temperature and salinity (). The Nha Trang central beach NB sample had the lowest number of isolates (8 with 6 yeasts and 2 filamentous fungi), while the deeper water DW sample expressed the highest number of filamentous fungi isolates (16 vs. 1–7 of all other samples) and none of the yeasts ().
Fungal isolates with morphological differences were further selected for DNA extraction, sequencing and identification based on ITS sequences. Previous studies have shown that some fungal species have at 99% and others at 93% their species-specific sequence similarity [Citation68]. To account for these differences, the UNITE community has clustered all ITS sequences at several sequence similarity thresholds to obtain approximate species-level OTUs referred to as species hypotheses with the 98.5% threshold level given [Citation60]. Moreover, a current systematic study suggested that the optimal identity threshold to discriminate filamentous fungal species is 99.6% for ITS [Citation69]. In the present study, among a total of 75 strains, 38/38 yeasts and 19/37 filamentous fungi were identified into species () with the fungal ITS rRNA gene sequences, which showed 99–100% identity with the closest related existing sequences of NCBI database (Table S1, Supplementary Material). All 18 other filamentous fungal strains were identified into genus (), including 4 sharing 95.6–98.3% identity with one closest related type strain (Group A) and 14 sharing 99–100% identity with at least two closest related type strains (Group B). Group A composes of DW7M (96.6% identity with Fusicolla acetilerea IMI 181488 (T)), DL11M (97.2% identity with Penicillium simplicissimum CBS 280.39 (T)), DW3M (98.16% identity with Stagonosporopsis oculi-hominis CBS 634.92 (T)), and DL12M (98.3% and 95.6% identity with Phanerochaete chrysosporium H008 and its type strain IFM 47473) (Table S1, Supplementary Material). Group B includes Cladosporium colombiae-like DM11M1 and NB8M; Aspergillus aflatoxiformans/austwickii-like TB8M, TB9M, DW1M and DW2M; A. costaricaensis-like TB10M; A. uvarum/japonicus-like DM11M2 and DM12M; A. versicolor-like VN17M, VN18M and HMB11M; Penicillium cuddlyae-like DW14M; and Trichoderma atrobrunneum/lixii-like SC13M (). The strains of Group A and some of Group B can be considered as potential novel species but require further studies with other marker genes and morphology (our unpublished data). One of these strains, DW14M, has currently been described as a novel species, Penicillium vietnamense sp. nov [Citation70], submitted in parallel to Mycobiology).
Table 2. Taxonomy of cultured fungal species found in this study.
3.2. Fungi diversity in the coastal ecosystems of Vietnam: Culture-independent approach
Three sampling sites HMC, CD, and DW showed a representatively high density of marine fungal community at Nha Trang Bay with differences in ecological characteristics and level of human impact on their habitats. Therefore, these sampling sites were selected for cultivated-independence studies. (1) HMC (Hon Mun Core Zone) is a representative of the core zone of Hon Mun marine protected ecosystems with low human impact, w/ere expressed the highest density of marine fungi at 105 CFU L−1 from the culture-dependent approach in this study. (2) CD (Cau Da Port), a harbor site with high-frequency human activity, is another worthy representative with the second-highest density of 5.4 × 104 CFU L−1. (3) DW (Deep waters at Nha Trang Bay) had the highest number of filamentous fungal strains isolated and identified, and also a high potential for the presence of uncultivable fungi in a different deeper habitat (lower pH and temperature but higher hydrostatic pressure) compared to surface coastal marine habitats as explained above.
From a total of 749,989 sequences after the assembly, a sum of 402,825 quality-filtered ITS reads were acquired after the expulsion of chimeric and the unique tag (Table S2, Supplementary Material). Subsequent to normalizing all data collections to the smallest sequence read and eliminating all uncommon taxa, the last analyzed data indexes contained 128 microeukaryotes OTUs. The noticed OTU wealth and Shannon variety were utilized straightforwardly for additional investigations. The abundance of total microeukaryotes from various samples was the highest at HMC (108 OTUs), followed by CD (70 OTUs) and lastly, DW (44 OTUs) (Table S2, Supplementary Material).
Among microeukaryote OTUs, fungi were relatively dominated with a total abundance of 13.82% (55 368 fungal read counts/400 639 microeukaryotes read counts) (Table S2, Supplementary Material). However, the richness of fungi was low with a total of 5 fungal OTUs ranging from 1 (DW sample) to 3 (CD and HMC samples) OTUs (). The majority of fungal OTUs were highly abundant: Candida_blankiiOTU_2 had a relative abundance of total fungi at 95.95% and 97.99% to CD and HMC samples, respectively, while the DW sample had 100% unclassified ChytridiomycotaOTU_101 (). The other frequently detected OTUs were Aspergillus_versicolorOTU_29 (1.67% at CD and 1.48% at HMC), Epicoccum_sorghinumOTU_65 (0.47% at HMC), and FungalOTU_104 (2.37% at CD and 0.05% at HMC).
Table 3. Phylogenetic affiliation and taxonomy of the fungal OTUs obtained with ITS primers based on cultivated-independence studies.
3.3. Comparison of culture-dependent and culture-independent approaches
The results of the fungi diversity and community composition of the 3 surfaces coastal marine and deeper waters (CD, HMC, DW) at Nha Trang Bay using culture-dependent and culture-independent approaches were similar to the most abundant phylum, genus and species but different at less common taxa. In particular, the phylum Ascomycota was found to be most common at 3 samples HMC, CD and DW with the average relative abundance of 89.50% and 99.68% from culture-dependent and culture-independent approaches, respectively (). At the genus level, only Candida and Aspergillus were detected in both approaches ( and Citation3). The results from both approaches showed that members of ascomycetes and these two genera were the most dominant in the mycoplankton community of Nha Trang Bay. Compared to the culture-dependent approach, the culture-independent approach additionally identified members of Chytridiomycota (ChytridiomycotaOTU_101) in the DW sample, other ascomycetes (Epicoccum_sorghinumOTU_65) in the HMC sample, and unclassified uncultured fungi (FungalOTU_104), where the sequence similarity was around 90% with the closest relative, in the CD and HMC samples.
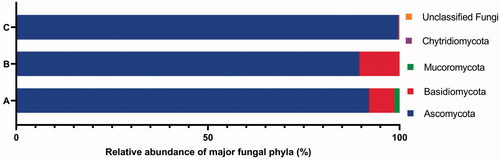
Figure 2. The percentage abundance of major phyla of fungi isolates from all 11 samples using culture-dependent approach (A), fungi isolates from 3 samples HMC, CD and DW using culture-dependent approach (B), and fungal OTUs from 3 samples HMC, CD and DW using culture-independent approach (C).
At the species level, Candida blankii was detected as the most abundant species in surface coastal marine habitats CD and HMC and absent in the deeper water DW in both approaches. In particular, the Candida blankii strains identified from the culture-dependent approach included 33.3% strains found in the CD sample, 75% strains in the HMC sample and none in the DW sample (), whereas the culture-independent approach measured the abundance of this species at 95.95%, 97.99% and 0.00% in the fungal community of samples CD, HMC and DW, respectively (). Interestingly, direct matching of the ITS sequences of fungi detected from these two approaches showed that Candida_blankiiOTU_2 in the CD and HMC samples from the culture-independent approach shared 99.65–100% identity (coverage 95%) to all 5 Candida blankii strains isolated from these two samples by the culture-dependent approach (), confirming the exact results from both methods.
3.4. Morphology, phylogeny and taxonomy of major fungal classes and orders
Regarding taxonomy, fungal taxa were identified using both morphology and molecular techniques. Identified species are listed in , morphology is presented in Supplementary Material (Figure S1), phylogenetic relationships in –Citation7 and checklist with additional information are provided in the Supplementary Material (Table S3). These results showed that the culturable fungal strains in this study belonged to 3 phyla, 5 subdivisions, 7 classes, 12 orders, 17 families, 22 genera and at least 40 taxa to species level, of which 29 species were identified. Hyphomucor assamensis (1.33%) was the only one identified as a member of the phylum Mucoromycota (formerly Zygomycota), whereas Ascomycota and Basidiomycota made up for 91.92% and 6.65% of total fungal strains, respectively (). Of the ascomycetes, strains belonged to 18 genera, including Penicillium and Aspergillus as the most diverse genera in species number (5-6 species each), followed by Candida (4 species), Cladosporium (3 species), Trichoderma (3 species), and other genera with one species each such as Aureobasidium, Stagonosporopsis, Alternaria, Cyphellophora, Parengyodontium, Simplicillium, Fusicolla, Nigrospora, Gelasinospora, Millerozyma, Meyerozyma, Pichia, and Kodamaea. Strains of Basidiomycota were affiliated with Rhodotorula (2 species), Rhodosporidiobolus, and Phanerochaete ().
Figure 3. Phylogenetic tree of Orders Capnodiales, Dothideales and Pleosporales (class Dothideomycetes, Phylum Ascomycota) based on ITS rRNA genes. The evolutionary history was inferred by using the Maximum Likelihood method and Kimura 2-parameter model [Citation54]. The tree with the highest log likelihood –2876.51) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 34.52% sites). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. This analysis involved 18 nucleotide sequences, including ones in this study (*), from type strains (T) and Chytridium olla UACCC ARG100 (T) used as an outgroup.
![Figure 3. Phylogenetic tree of Orders Capnodiales, Dothideales and Pleosporales (class Dothideomycetes, Phylum Ascomycota) based on ITS rRNA genes. The evolutionary history was inferred by using the Maximum Likelihood method and Kimura 2-parameter model [Citation54]. The tree with the highest log likelihood –2876.51) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 34.52% sites). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. This analysis involved 18 nucleotide sequences, including ones in this study (*), from type strains (T) and Chytridium olla UACCC ARG100 (T) used as an outgroup.](/cms/asset/e10f4bfd-6b26-4274-8ce9-4164356bb59c/tmyb_a_2008103_f0003_b.jpg)
Figure 4. Phylogenetic tree of Orders Chaetothyriales and Eurotiales (Class Eurotiomycetes, Phylum Ascomycota) based on ITS rRNA genes. The evolutionary history was inferred by using the Maximum Likelihood method and Kimura 2-parameter model [Citation55]. The tree with the highest log likelihood (–3375.53) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.3870)). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. This analysis involved 41 nucleotide sequences, including ones in this study (*), from type strains (T) and Chytridium olla UACCC ARG100 (T) used as an outgroup.
![Figure 4. Phylogenetic tree of Orders Chaetothyriales and Eurotiales (Class Eurotiomycetes, Phylum Ascomycota) based on ITS rRNA genes. The evolutionary history was inferred by using the Maximum Likelihood method and Kimura 2-parameter model [Citation55]. The tree with the highest log likelihood (–3375.53) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.3870)). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. This analysis involved 41 nucleotide sequences, including ones in this study (*), from type strains (T) and Chytridium olla UACCC ARG100 (T) used as an outgroup.](/cms/asset/ecb9affc-d949-4b1c-b9dc-e8df0ce144cc/tmyb_a_2008103_f0004_b.jpg)
Figure 5. Phylogenetic tree of Orders Hypocreales, Trichosphaeriales and Sordariales (Class Sordariomycetes, Phylum Ascomycota) based on ITS rRNA genes. The evolutionary history was inferred by using the Maximum Likelihood method and Kimura 2-parameter model [Citation54]. The tree with the highest log likelihood (–3297.12) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.3655)). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. This analysis involved 18 nucleotide sequences, including ones in this study (*), from type strains (T) and Chytridium olla UACCC ARG100 (T) used as an outgroup.
![Figure 5. Phylogenetic tree of Orders Hypocreales, Trichosphaeriales and Sordariales (Class Sordariomycetes, Phylum Ascomycota) based on ITS rRNA genes. The evolutionary history was inferred by using the Maximum Likelihood method and Kimura 2-parameter model [Citation54]. The tree with the highest log likelihood (–3297.12) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.3655)). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. This analysis involved 18 nucleotide sequences, including ones in this study (*), from type strains (T) and Chytridium olla UACCC ARG100 (T) used as an outgroup.](/cms/asset/0fe01a79-dc9b-45f6-9310-4f6debbefaf9/tmyb_a_2008103_f0005_b.jpg)
Figure 6. Phylogenetic tree of Order Saccharomycetales (Class Sacharomycetes, Phylum Ascomycota) based on ITS rRNA genes. The evolutionary history was inferred by using the Maximum Likelihood method and Tamura 3-parameter model [Citation54]. The tree with the highest log likelihood (–3834.69) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Tamura 3 parameter model, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.5029)). This analysis involved 42 nucleotide sequences, including ones in this study (*), from type strains (T) and Chytridium olla UACCC ARG100 (T) used as an outgroup.
![Figure 6. Phylogenetic tree of Order Saccharomycetales (Class Sacharomycetes, Phylum Ascomycota) based on ITS rRNA genes. The evolutionary history was inferred by using the Maximum Likelihood method and Tamura 3-parameter model [Citation54]. The tree with the highest log likelihood (–3834.69) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Tamura 3 parameter model, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.5029)). This analysis involved 42 nucleotide sequences, including ones in this study (*), from type strains (T) and Chytridium olla UACCC ARG100 (T) used as an outgroup.](/cms/asset/2c645ec5-8c91-4cf3-a8e9-422cd891b239/tmyb_a_2008103_f0006_b.jpg)
Importantly, 12 out of 29 identified fungal species were first reported with a marine origin (Table S3, Supplementary Material). Moreover, 17 identified species have not been reported in a neighboring sea area as China (Table S3, Supplementary Material). Only Candida tropicalis was found from oil-polluted sediments collected in coastal zones of Vietnam [Citation71]; and thus other 28 identified species detected in this research were new records in a marine ecosystem of Vietnam (Table S3, Supplementary Material).
Interestingly, Candida was the most abundant genus with 25 strains isolated in all 10 surface coastal marine habitats. Candida blankii was the most abundant species with 15 strains originating from all surface coastal marine habitats, except 2 beaches DL and NB. The deeper waters DW generally shared a common marine fungal community composed of major genera Aspergillus, Penicillium and Cladosporium with surface coastal marine habitats. However, DW did not have Candida or any basidiomycetes. The presence of Parengyodontium, Simplicillium, Fusicolla, and Stagonosporopsis in DW was also different from fungal communities in the surface marine habitats (; Table S2, Supplementary Material).
3.5. Enzyme activity of marine fungal strains
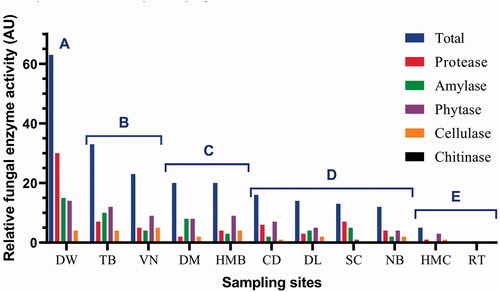
We investigated extracellular enzyme activity including protease, amylase, phytase, cellulase and chitinase of all 75 fungal strains obtained from a culture-dependent approach. The results are shown in Table S4 (Supplementary Material). The total relative extracellular enzyme activity (AU) of fungal strains from each sampling site was calculated, in which 1 AU was defined as an enzyme activity unit with hydrolysis zone diameter D–d = 10–15 mm, 2 AU for D–d = 15–20 mm and 3 AU for D–d > 20 mm. As a result, the total relative enzyme activity of each sample ranged from 0 AU in RT to 63 AU in DW (). Based on total relative enzyme activity, the samples were divided into 5 groups. Interestingly, each group displayed a specific habitat or ecological niche. Group A (>60 AU) was for deep waters, Group B (23–33 AU) for major aquaculture regions, Group C (20 AU) as a buffer region with some aquaculture and some tourism activity, Group D (12–16 AU) for major tourism regions (beach bathing, anchoring, fishing, and tourism boat running), and Group E (0–5 AU) for marine protected area cores with very limited human activity ().
Figure 7. Phylogenetic tree of Order Polyporales (Class Agaricomycetes, Phylum Basidiomycota), Order Sporidiobolales (Class Microbotryomycetes, Phylum Basidiomycota) and Order Mucorales (Class Mucoromycetes, Phylum Mucoromycota) based on ITS rRNA genes. The evolutionary history was inferred by using the Maximum Likelihood method and Tamura 3-parameter model [Citation54]. The tree with the highest log likelihood (–3580.61) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Tamura 3 parameter model, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.8620)). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. This analysis involved 14 nucleotide sequences, including ones in this study (*), from related type strains (T) and Chytridium olla UACCC ARG100 (T) used as an outgroup.
![Figure 7. Phylogenetic tree of Order Polyporales (Class Agaricomycetes, Phylum Basidiomycota), Order Sporidiobolales (Class Microbotryomycetes, Phylum Basidiomycota) and Order Mucorales (Class Mucoromycetes, Phylum Mucoromycota) based on ITS rRNA genes. The evolutionary history was inferred by using the Maximum Likelihood method and Tamura 3-parameter model [Citation54]. The tree with the highest log likelihood (–3580.61) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Tamura 3 parameter model, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.8620)). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. This analysis involved 14 nucleotide sequences, including ones in this study (*), from related type strains (T) and Chytridium olla UACCC ARG100 (T) used as an outgroup.](/cms/asset/c4fbb808-b2f6-44d1-a169-86a5766ba024/tmyb_a_2008103_f0007_b.jpg)
Figure 8. Relative extracellular enzyme activity (AU) of all fungal strains from each sample at Nha Trang Bay and Van Phong Bay, Khanh Hoa province, Vietnam. For each strain, 1 AU was defined as an enzyme activity unit with hydrolysis zone diameter D–d = 10–15 mm, 2 AU for D–d = 15-20 mm and 3 AU for D–d > 20 mm. Based on total relative enzyme activity shown, the samples were divided into 5 groups: Group A (>60 AU), Group B (23–33 AU), Group C (20 AU), Group D (12–16 AU), and Group E (0–5 AU).
3.6. A checklist of trophic mode and guild analyses of marine fungal strains and OTUs
A checklist of trophic mode and guild analyses of all fungal strains and OTUs identified from the culture-dependent and culture-independent approach in this study, with their possible or confirmed functions, are presented in Table S3, Supplementary Material. The results suggest that marine fungi living in Nha Trang Bay and Van Phong Bay can exist in numerous life modes, such as saprotrophs, endophytes, symbionts, or pathogens to humans, animals, and plants. For example, Candida yeasts, which were most frequently detected, can form symbiotic relationships with plants and can be a pathogen to humans and animals. Aspergillus versicolor-like group can be saprotrophs with aliphatic and aromatic hydrocarbon degradation activity.
4. Discussion
4.1. Fungi biodiversity, community composition and enzyme activity in the coastal marine ecosystems in Vietnam
This is the first study comprehensively exploring the fungal diversity using culture-dependent and culture-independent approaches of the coastal marine habitats of Nha Trang Bay and Van Phong Bay, Khanh Hoa province in Vietnam. These habitats are experiencing rapid anthropogenic disturbances from urbanization, tourism, shipping and aquaculture expansion [Citation6,Citation43–45,Citation72,Citation73] which are predicted to seriously affect marine habitats. We have retrieved fungal ITS sequences belonging to Ascomycota, Basidiomycota, and some species/genera of Mucoromycota, Chytridiomycota and unclassified and/or uncultured fungi specimens. At the species level, 12, 17 and 28 out of 29 identified fungal species have been not reported in a marine environment, a sea area of China and a sea area of Vietnam, respectively. Therefore, these new records contribute to discovering fungal diversity in the global, regional and national coastal marine habitats.
The present study shared 3 common fungal phyla (Ascomycota, Basidiomycota and Mucoromycota) and 8 common fungal genera (Aspergillus, Trichoderma, Pleosporales (currently Stagonosporopsis and Alternaria), Aureobasidium, Candida, Rhodotorula and Rhodosporidiobolus (Rhodosporidium)) to culturable fungi composition in the coastal waters of Southern China [Citation26]. Similarly, a culture-independent study on the diversity of planktonic fungi in the coastal waters of the Bohai Sea in China detected more than 60% of unclassified fungi, the dominance of Ascomycota (20.82%), Basidiomycota (9.72%) and Chytridiomycota (2.64%), and episodic low dominance of Cryptomycota (0.64%) and Mucoromycota (0.18%) [Citation27]. Our results showed a super-dominance of Ascomycota (99.68%), followed by Chytridiomycota (0.16%) and unclassified fungi (0.16%). While diverse taxa with 283 genera within Ascomycota were discovered in the Bohai Sea [Citation27], only 5 fungal OTUs were found at Nha Trang Bay in our study. Several previous studies have suggested that mycoplankton can use dissolved organic carbon [Citation74,Citation75], thus, fungal diversity can be attributed to the lower carbon from autochthonous primary production and terrestrially-derived production in the coastal region at Nha Trang Bay than that in the Bohai Sea [Citation23,Citation40,Citation27].
Molecular diversity analysis of marine fungi across 130 European environmental samples also shows the widespread of Candida yeasts and two major genera of Basidiomycota yeasts including Rhodotorula and Rhodosporidium [Citation28], which are similar to our findings. These results suggest that a yeast lifestyle of Dikarya (Ascomycota and Basidiomycota) was the highly dominant fungi in marine ecosystems and adapt well to these environments [Citation37,Citation76]. Especially, many yeast morphotypes detected are closely related to opportunistic animal pathogens, as shown in Table S3, Supplementary Material, suggesting that they may be parasites or opportunistic pathogens of marine animals. We do not exclude the possibility that some filamentous fungi may not be fully recovered from water filtration and/or DNA/RNA extraction [Citation28]. For example, in the present study, Pezizomycotina, or filamentous ascomycetes, were recovered at very low abundance with an exception to Aspergillus which appeared at higher abundance in both cultivation and molecular approaches ( and Citation3).
Importantly, our results showed that fungi diversity and community composition of the surface coastal marine and deeper waters differed across sampling sites, possibly reflecting the different impact levels from human activities and ecological factors in the marine environment in Khanh Hoa, Vietnam. For example, the lowest density of fungi (102 CFU L−1) was found in the samples NB and DL where both locations are beaches with an extremely high level of tourism activity. As expected, the highest density of fungi (105 CFU L−1) was found in the Hon Mun Marine Protected Area of Nha Trang Bay where the human impact on this area is minimal. These findings suggest that human activity can negatively impact the biodiversity of marine organisms, including fungi, and such marine protected areas established are likely essential to the life of many marine fungi. For example, some sunscreen lotions and personal-care products contain benzophenone-3, which are highly toxic to corals [Citation77] and the negative effect of this substance on marine fungi is in question. Moreover, the deep water DW had a relatively low density of fungi (8.8 × 102 CFU L−1) but had the highest number of filamentous fungi isolates (16 vs. 1–7 of all other samples) and none of the yeasts were successfully cultivated, which indicated the highest diversity of culturable filamentous fungi and maybe suggested uncultivable fungi existing in such a unique deep habitat that differs from surface sea habitats with lower temperature and higher hydrostatic pressure [Citation10].
The impact of human and ecological factors on the biodiversity of marine fungi at Nha Trang Bay and Van Phong Bay was also displayed via the total relative enzyme activity of fungi isolates in each sampling site. As shown in , five groups of similarly ecological sampling areas were given, which well explained the correlation between human impact and dissolved organic matter concentration in marine ecosystems. Group A of deep waters at Nha Trang Bay expressed the highest total relative enzyme activity and also the highest activity of protease, amylase and phytase. This can involve a high diversity of fungal isolates and also the availability of organic matters of carbon, nitrogen and phosphorous, in which carbon sources can be sunk from the surface euphotic zone. Group B with the second-highest total enzyme activity and the highest cellulase activity typically represented for environments enriched with cellulose and other organic substrates in major aquaculture regions at Nha Trang Bay and Van Phong Bay. Group C displayed a buffer region with some aquaculture activity and some tourism activity, thus an average number in total relative enzyme activity was recorded. In Group D, tourism activities such as swimming, anchoring, fishing and operating by tourism boats can affect the mycoplankton and phytoplankton diversity and sink organic matters to a lower abundance, thus can lower the enzyme activity of fungi. Finally, Group E for marine protected area cores with very limited human activity displayed a very low or no enzyme activity of fungal isolates, which suggested other enzyme activity or unknown ecological functions of fungal communities living in these ecosystems. Although further direct evidence about the impact of human and ecological factors on fungal biodiversity at Nha Trang Bay and Van Phong Bay is necessary, the results from the present study provide first and valuable clues about their effects.
4.2. Comparison of culture-dependent and culture-independent approaches
The total community of marine fungi, especially uncultured fungi, in the sea areas of Vietnam is not known. We fill this voids by focusing on the planktonic fungi at 3 sampling locations of Nha Trang Bay, Khanh Hoa province, Vietnam using both culture-dependent and culture-independent approaches. Our findings revealed that Candida blankii yeasts are the most dominant fungi species at Nha Trang Bay and this result was consistent regardless of the use of the two methods. However, the abundance of other taxa depended on which method was used. In the culture-dependent method, at least 40 species belonging to 22 genera within 3 phyla Ascomycota, Basidiomycota and Mucoromycota were found (), while in the culture-independent method, only 5 fungal OTUs were uncovered, in which 3 identified OTUs belonged to 3 different genera within Ascomycota and 2 unidentified OTUs within Chytridiomycota and unclassified environmental clusters ().
Even though isolated directly from the sea water samples, some fungi were not able to grow on culture media. For example, fungal isolates were obtained on all five different media (MEA, SDA, CMA, PDA and CMA) during isolation from deep-sea sediments [Citation42], whereas only two (MEA and SDA) of these five media were found to recover marine fungi from the coastal waters [Citation26]. In the later case, SDA was found to be a better medium than MEA for the isolation of marine fungi, although there was no statistical significance [Citation26]. In the present study, 75 strains of 3 major phyla Ascomycota, Basidiomycota and Mucoromycota were isolated on SDA, but none of the isolates from the phylum Chytridiomycota were recovered on this medium as found from our metabarcoding analysis. Although chytrids (Chytridiomycota), or “early-diverging lineages” of fungi, have been found to dominate nearshore and sediment samples [Citation28,Citation78], our results suggest that the diversity of chytrids is highly likely based on environmental sequencing data [Citation79].
However, there are several constraints to the culture-independent approach. In the present study, ascomycetes were found to dominate from culture-dependent and culture-independent approaches but for the latter method, it is difficult to detect members of Basidiomycota and Mucoromycota that are present on the SDA medium. The reason for this is that the relative abundance of microbial species in a natural habitat is rarely equal. DNA may not be recovered from all genotypes and the results of next-generation sequencing, which are mainly based on analysis of ITS regions for fungi [Citation80], can be biased toward the most abundant organisms at the time of sampling [Citation81]. Moreover, the ITS rDNA region primers were designed using genomic regions from mainly terrestrial representatives which were biased toward terrestrial Dikarya (Basidiomycota and Ascomycota), resulting in poor representation of marine fungi taxa [Citation9].
The sequencing-based on the fungal ITS rDNA region even expressed both advantages and disadvantages. In the present study, it can coamplify other microeukaryotes and these eukaryotes are reported that they often dominate marine environmental metabarcoding sequence data, resulting in limited representation by marine fungi [Citation9]. Fortunately, ascomycetes were still found to dominate other eukaryotes in the HMC sample, as shown in in this study.
Another disadvantage of the culture-independent approach is that the correspondence of OTU with species can be inconsistent. OTUs are defined dependent on the identity, typically with 97% [Citation82]. However, some species have genes that are 97% identity, which will bring about merged OTUs containing various species. Additionally, a solitary species may have paralogs that are 97% identity, making the species be part across at least two species. Some or even many identified clusters might be wrong because of the artifacts, including reading errors and chimeras [Citation82]. For a long time, sequencing of household and other genes has been utilized exclusively [Citation83], but then a polyphasic approach to taxonomy based on morphological, ecophysiological and molecular analyses appeared to be more effective to discover species [Citation84]. Currently, all-important phenotypic features can be used for classification in agreement with molecular features, e.g., a case of study on Aspergillus and Penicillium [Citation85]. In the present study, to increase the reliability of species identification compared to the use of ITS1-F and ITS4 primers, DNA barcoding targeting the entire ITS region using ITS1-F_KYO2 and ITS4 primers was designed. This was based on the previous evidence that ITS1-F_KYO2 and ITS4 matched around 99% within the ascomycetes, basidiomycetes, or “non-Dikarya” group [Citation48]. Although the primers ITS1-F_KYO2 and ITS4-R were used to improve coverage across diverse taxonomic groups of fungi compared to previously existing primers [Citation48], these primers may not amplify ITS regions from all marine fungi in our study.
Finally, the culture-independent approach could not distinguish metabolically inactive fungi from true marine fungi as cultivated on media [Citation9]. Thus, this can make it difficult to compare the abundance from both methods and requires additional lines of evidence to capture and characterize marine fungi cells living in their ecosystems. Therefore, the combined use of culture-dependent and culture-independent approaches in the present study may provide a better characterization of marine fungi composition than that obtained when using each approach independently. Most importantly, the direct matching of the ITS sequences of fungi detected from these two approaches has confirmed that molecular analyses are in agreement with phenotype features of fungal isolates.
4.3. Comparison of surface coastal marine and deeper (transition layer) waters
We found consistent evidence that Aspergillus, Penicillium and Cladosporium are dominant genera in both surfaces coastal marine and deeper waters (a transition layer rather than real deepsea) at Nha Trang Bay, Vietnam. Further, we also identified a number of unique and self-contained niches for fungi in such marine habitats. Coastal regions are characterized by eutrophication from terrestrial run-off and high primary production [Citation86]. In contrast, the deepsea layers have the salient features of low temperatures, high hydrostatic pressure, absence of light, and low biological diversity [Citation10]. This leads to higher availability of organic matter as detritus in the coastal marine habitats compared to deepsea regions. Thus, in the coastal waters, prokaryotic heterotrophs are known to play a massive role as primary degraders, followed by eukaryotic heterotrophs, including micro-sized animals and marine fungi [Citation87].
Compared to the coastal marine habitats, the deepsea sediments expressed a very low species richness [Citation41,Citation88], and the majority of the fungi belonged to Ascomycota and Basidiomycota with a large representation of yeasts belonging to both the phyla [Citation37,Citation40]. Molecular studies have revealed the presence of the additional major fungal phyla Chytridiomycota and Zygomycota [Citation10]. In the present study, culturable fungi from deeper waters DW at a depth of 40–47 m, even though not really deepsea, had a relatively low density at 102 CFU L−1 compared to higher densities ranging from 102 to 105 CFU L−1 of samples in the surface sea habitats, confirming a low fungal richness in the deep habitats as known from previous studies on deepsea. However, DW displayed the dominant number of filamentous fungi isolates and none of the yeasts. It had been previously suggested that yeast forms dominate fungal diversity in the deep-water column and marine sediment environments [Citation37,Citation40]. Therefore, a dominant representation of filamentous fungi in DW suggested that DW could be a “transition layer” to sink and likely reduce fungal filaments from the surface sea to deepsea layers. Moreover, the results showed a wide diversity of fungal phenotypes in different marine fungal communities and were consistent with their molecular diversity analysis [Citation76].
4.4. The first record of Candida blankii yeast as a dominant fungi species in marine habitats
Although Candida yeast has been found as one of the most widespread fungi species in marine habitats, especially in a big-sized molecular analysis across 130 European marine samples [Citation28], Candida blankii has been detected only in a few locations in Japan, Sweden, New Zealand and Canada (https://eol.org/pages/6770416). This study presents the first record of Candida blankii as a dominant species in a marine habitat in Vietnam. This yeast species was detected on SDA medium with 15 isolates of total of 75 fungi isolates (20%) originated from all surface coastal marine habitats at Nha Trang Bay and Van Phong Bay with exceptions from 2 beaches DL and NB only. Similarly, next-generation sequencing method showed its relative abundance to total fungi at 95.95% and 97.99% in CD and HMC samples at Nha Trang Bay, respectively. Moreover, 10 isolates of other Candida species were found on SDA medium, which belonged to C. tropicalis, C. akabanensis and C. intermedia. Collectively, a third of culturable fungi isolates in marine areas of Khanh Hoa province, Vietnam was identified as Candida yeasts. Notably, none of Candida yeasts were recorded in the deep water DW in both culture-dependent and culture-independent approaches.
Candida is a heterogeneous genus of budding yeast (Saccharomycotina) in the family Saccharomycetaceae and is estimated to constitute 25% of all yeast species. It comprises primarily species related to plants, spoiling vegetation, and plant-feeding insects [Citation89]. The level of Candida species can utilize plant materials such as xylose, cellobiose, aliphatic hydrocarbons and starch is reported as 71%, 57%, 29% and 27%, respectively, while 85% of species require vitamins delivered mostly in plant biomass. However, 65% of Candida species cannot grow at 37 °C or higher temperatures, therefore just generally a couple of species can infect humans and animals [Citation89].
C. blankii is a rare and uncommon yeast but can form symbiotic relationships with plants [Citation90] or be considered as an opportunistic human pathogen [Citation91]. For example, it was shown as the most common species colonizing flower nectaries in India and making Azadirachta indica flower more, although this relationship is not fully understood yet [Citation90]. Regarding human infections, C. blankii was first described from the blood of a mink in 1968 [Citation92] and later described from clinical specimens, including the bloodstream of a fungaemia patient [Citation93,Citation94]. Although a few cases of C. blankii have been found, other rare and new species of fungi related to human infection have been frequently described in recent years [Citation95,Citation96]. Candida species are the third most common cause of sepsis in a recent prevalence survey of adults and children hospitalized in the United States [Citation97]. The outbreaks of fungal sepsis due to emerging and rare Candida species, particularly multidrug-resistant Candida auris have received great attention from the medical community [Citation95,Citation98]. Currently, a report has emphasized the emergence of C. blankii with reduced susceptibility to one or more antifungal agents in nosocomial fungaemia [Citation96].
Moreover, C. blankii can grow at elevated temperatures and high salt concentrations, and its genome analysis suggested a strongly altered 1,3-beta-D-glucan synthase protein sequence, which may help it occur in hydrocarbon-rich environments [Citation96,Citation99]. This yeast is studied extensively for use in xylose fermentation and the degradation of hemicellulose hydrolycates. These facts explain why it has appropriate attributes to be a colonizer in surface coastal marine habitats with high temperature and salinity as monitored at the sampling areas in the present study. This was also consistent with evidence that no Candida yeasts were detected in the deep water DW with a temperature of 22 °C and higher hydrostatic pressure. In fact, other Candida species are also those impacted when grown under marine conditions. For example, a marine yeast Candida tropicalis BH-6 isolated from mangrove sludge had the optimum temperature for growth at 37 °C, the initial pH value at 5.0, and the wide NaCl concentrations of 0–6% [Citation100]. Candida oceani expressed the optimal growth at 3% sea salt [Citation101] and Candida viswanathii displayed filamentous morphology under elevated hydrostatic pressure [Citation38].
In Vietnam, the yeasts C. viswanathii and C. tropicalis were found from oil-polluted sediments collected in coastal zones with transformation activity of branched aromatic hydrocarbons in biofilm [Citation71,Citation102], while C. tropicalis was detected as the most common Candida species from blood cultures in two hospitals between May 2013 and May 2015 [Citation103]. Moreover, C. tropicalis, Diutina rugosa and Pichia kudriavzevii were detected in traditional alcoholic fermentation in Vietnam [Citation104]. In the present study, the dominance of 15 C. blankii isolates from all sampled marine habitats with an exception at 2 beaches of Nha Trang Bay and Van Phong Bay, where human contact to seawater occur most frequently and directly, suggests that these isolates are unlikely human pathogens or from a clinical source. It is more likely that C. blankii plays a primary role as a hydrocarbon transformer in marine ecosystems as discussed above. A current study has shown that urban and tourism activities are typical pollution sources of polycyclic aromatic hydrocarbons and other organic contaminants in the coastal areas in Spain [Citation105]. The question is that whether the increasing activities from urbanization, tourism, shipping and aquaculture expansion at Nha Trang Bay and Van Phong Bay in Vietnam [Citation7,Citation43,Citation44,Citation73] can give us a clue on the dominance of C. blankii species or not. Therefore, the biological nature and ecological role of this species in association with changing environmental conditions in this region should be further studied, including hydrocarbon transformation activity, human pathogenicity, and originality in terms of marine or clinical sources, or a transition from terrestrial to marine habitats.
4.5. Potential ecological role of mycoplankton in the marine food web
In spite of the fact that the diversity and ecological function of marine fungi have generally been unexplored, some ongoing cultivation and metabarcoding studies of microorganisms in the coastal marine environments and relationship with macroeukaryotes have indicated that fungi are a dominant kingdom and play a significant part in the marine nutrition cycles [Citation9,Citation10]. Marine fungi, especially filamentous fungi, enable to break down organic matters, basically carbohydrates and polyphenols of plant and algae. The plant and algae biomass is the most renewable energy feedstock on the Earth and induces the biosynthesis of microbial extracellular enzymes to catalyze their cleavage into shorter sugars [Citation14,Citation29]. In this manner, marine fungi have generally been found in the ocean, especially connected with sediment, seawater, submerged plant, and algae. Fungi are hypothesized to contribute to phytoplankton population dynamics, biological carbon pump and change in marine sediments [Citation9]. They can act as predators, pathogens and parasites, and can be discovered living in a symbiotic relationship with plants, algae, animals and other organisms [Citation106]. Regardless of the different roles of marine fungi, their ecological functions are still open questions and unsolved issues.
Based on the roles of fungi in the marine carbon cycle as described by [Citation9], the checklist of trophic mode and guild analyses (Table S3, Supplementary Material), and total relative enzyme activity (), we hypothesized a contribution of plankton fungi in 3 locations of the study (HMC, CD, DW) at Nha Trang Bay as follows. Many saprotrophic fungi (mainly Candida, Aspergillus, Penicillium and Cladosporium) can use particulate and dissolved organic carbon from phytoplankton, which is then fed by zooplanktons in the marine food web. Some other fungi can be pathogens, endophyte, or epiphyte to human, animal, plant and algae.
Identifying the role of marine saprotrophic fungi in ecological cycles remains a key challenge as it does not simply detect their presence but also explores their activity. This can be addressed partly through the activity of extracellular enzymes they produce. Fungal strains isolated from different substrates in marine environments are producers of hydrolytic and/or oxidative enzymes such as alginate lyase, amylase, cellulase, chitinase, glucosidase, inulinase, keratinase, ligninase, lipase, nuclease, phytase, protease, and xylanase [Citation107]. In the present study, all fungal isolates were investigated with major extracellular enzymes, including protease, amylase, cellulase, phytase and chitinase. The activity of fungal isolates confirmed the roles of saprotrophic mycoplankton in the biogeochemical cycles of carbon, nitrogen and phosphorous. In particular, protease plays an important role in the biogeochemical cycle of nitrogen, whereas amylase and cellulase display a similar function in the biogeochemical cycle of carbon. Phytase, which catalyzes the release of phosphate from phytate (myco-inositol-hexaki-phosphate), plays an essential role in the biogeochemical cycle of phosphorous. Besides, as chitin is a component of the cell walls of fungi and exoskeletal elements of some animals (mollusks and arthropods), some fungi possess degradative chitinases related to their role as detritivores and also to their potential as arthropod pathogens. In our study, none of the chitinase activity was found in all fungal isolates suggested that mycoplankton do not seem to relate to the degradation of chitin.
Our study also provides strong evidence of phytoplankton-derived organic matter consumption by marine saprotrophic mycoplankton. Fungal isolates from this study with amylase and cellulase activity, as shown in , suggesting their potential to degrade phytoplankton polysaccharides. Moreover, species of Cladosporium, which were found in surface marine coastal and deeper waters in the present study, are considered as active saprotrophs in coastal waters and directly assimilate phytoplankton organic carbon. This is because Cladosporium is found to secrete the extracellular enzyme glucan 1,3-β-glucosidase [Citation108]. This enzyme can digest phytoplankton-derived organic matter and its abundance, in particular, correlates with increased abundances of specific diatom species and in the deep chlorophyll maxima regions of the oceans where phytoplankton biomass can be the highest. These results indicate some marine mycoplankton have a saprotrophic functional role in processing phytoplankton polysaccharides.
Although not directly linked to enzyme activity, the dominant presence of some groups of filamentous fungi such as Cladosporium and Aspergillus are among those known to participate in aliphatic hydrocarbon degradation, while the genera Penicillium and Aspergillus are among those known to take part in the degradation of aromatic hydrocarbons [Citation109–111]. Fungi have a relatively high tolerance to hydrocarbons [Citation109] and are likely primary degraders of high-molecular-weight hydrocarbons via secreted extracellular enzymes and work synergistically with other microorganisms in the oil degradation [Citation112]. Similarly, the yeasts Candida viswanathii and C. tropicalis isolated from oil-polluted sediments in Vietnam expressed the transformation activity of branched aromatic hydrocarbons [Citation71,Citation102].
Therefore, the dominance of Candida, Aspergillus, Penicillium and Cladosporium in the present study suggested the contribution of marine saprotrophic ascomycetes fungi to the degradation of oil and other hydrocarbons as organic matter from phytoplankton. The primary function of fungi, including mycoplankton is to degrade detrital organic matter from phytoplankton, which acts as primary producers and provides food for consumers at higher trophic levels. By interacting with microbial communities, mycoplankton productively converts particulate organic matter to dissolved organic matter as part of the biogeochemical cycles. Along with heterotrophic bacteria, mycoplankton play significant functions in the biogeochemical cycles of carbon, nitrogen, phosphorous, and other nutrient fluxes in marine ecosystems. It has been demonstrated that there are higher densities of mycoplankton close to the surface and in shallow waters, proposing their connection to the rise of organic matter. This keeps on connecting with crowded phytoplankton communities at the surface, inferring that mycoplankton is firmly identified with the utilization of organic matter in the euphotic zone [Citation9].
The functional roles of marine mycoplankton are poorly understood, resulting in a lack of understanding of their ecology. Due to the presence of bacterioplankton in the same niche, mycoplankton in the interactions with phytoplankton to transfer particulate organic carbon in marine food webs, therefore, maybe need to be further characterized. Our findings and previous studies suggest that marine fungi can contribute to the action of the biological carbon pump, which transfers the organic matter produced by phytoplankton via photosynthesis from the surface euphotic zone to sink as “marine snow” through the epipelagic, mesopelagic and bathypelagic zones to the deep ocean [Citation113]. The significance of fungal biomass in the marine ecosystem, therefore, requires further analysis, especially in terms of the current view that prokaryotes are the principal contributors of heterotrophic microbial biomass in the ocean [Citation114].
5. Conclusions
Fungi diversity and community composition of the surface coastal marine and deeper waters of Khanh Hoa province in Vietnam were uncovered using both culture-dependent and culture-independent approaches. Using a culture-dependent approach, the fungal community of 11 surfaces coastal marine and deeper waters at Nha Trang Bay and Van Phong Bay accounted for from 102 CFU L−1 to 105 CFU L−1 with a total of 75 strains, including 38 yeasts and 37 filamentous fungi successfully isolated and identified based on ITS sequences into species or genus. They belong to 3 phyla, 5 subdivisions, 7 classes, 12 orders, 17 families, 22 genera and at least 40 species. Among them, 29 species have been identified and many potential novel species need to be further studied; 12 and 28 species detected in this research are new records in a global marine source and a sea area of Vietnam, respectively. Using cultivated-independence studies on three representative samples HMC, CD and DW at Nha Trang Bay, the results showed that the marine fungal community displayed a dominance of Ascomycota and a lower abundance of some members of Chytridiomycota and unclassified uncultured fungus, but the richness of fungi was low with 5 fungal OTUs only. The results from both approaches shared similar fungal taxonomy to the most abundant phylum (Ascomycota), genera (Candida and Aspergillus) and species (Candida blankii) but were different at less common taxa. This is the first record of C. blankii as a dominant species in a marine area in Vietnam.
Major extracellular enzymes including protease, amylase, cellulase, phytase and chitinase of all fungi isolates were investigated. It suggested the roles of mycoplankton in the biogeochemical cycles of carbon, nitrogen, and phosphorous. Chitinase activity was not found in any mycoplankton. Based on the total relative enzyme activity, five groups of similarly ecological sampling areas were created, which was well explained the correlation to human impact and dissolved organic matter concentration in marine ecosystems. The checklist of marine fungal isolates and OTUs identified from our research indicated that they may be mostly saprotrophs (mainly Candida, Aspergillus, Penicillium and Cladosporium), commensals, or pathogens of human, marine animals, plants and algae.
Supplemental Material
Download MS Word (2.6 MB)Acknowledgments
The authors would like to acknowledge Tran Chau Loan, Nguyen Minh Tan and Huynh Ngo Y Nhi at Nha Trang University (NTU) for technical and sampling support, and Prof. Doan Nhu Hai at Institute of Oceanography (IO), Vietnam for deepwater sampling support.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Selig ER, Turner WR, Troëng S, et al. Global priorities for marine biodiversity conservation. PLoS One. 2014;9(1):e82898.
- Yao Y, Wang J, Yin J. Marine heatwaves in China’s marginal seas and adjacent offshore waters: past, present, and future. J Geophys Res Oceans. 2020;125:e2019.
- Dinh KV. Vietnam’s fish kill remains unexamined. Science. 2019;365:333.
- Halpern BS, Frazier M, Afflerbach J, et al. Recent pace of change in human impact on the world’s ocean. Sci Rep. 2019;9(1):11609.
- Jambeck JR, Geyer R, Wilcox C, et al. Marine pollution. Plastic waste inputs from land into the ocean. Science. 2015;347(6223):768–771.
- Nghia ND, Lunestad BT, Trung TS, et al. Heavy metals in the farming environment and in some selected aquaculture species in the Van Phong Bay and Nha Trang Bay of the Khanh Hoa province in Vietnam. Bull Environ Contam Toxicol. 2009;82(1):75–79.
- Nguyen X-V, Tran M-H, Le T-D, et al. An assessment of heavy metal contamination on the surface sediment of seagrass beds at the Khanh Hoa Coast, Vietnam. Bull Environ Contam Toxicol. 2017;99(6):728–734.
- Dinh KV, Nguyen QTT, Vo T-M-C, et al. Interactive effects of extreme temperature and a widespread coastal metal contaminant reduce the fitness of a common tropical copepod across generations. Mar Pollut Bull. 2020;159:111509.
- Amend A, Burgaud G, Cunliffe M, et al. Fungi in the marine environment: Open questions and unsolved problems. mBio. 2019;10(2):e01189–18.
- Manohar CS, Raghukumar C. Fungal diversity from various marine habitats deduced through culture-independent studies. FEMS Microbiol Lett. 2013;341(2):69–78.
- Worm B, Barbier EB, Beaumont N, et al. Impacts of biodiversity loss on ocean ecosystem services. Science. 2006;314(5800):787–790.
- Zaky AS, Greetham D, Louis EJ, et al. A new isolation and evaluation method for marine-derived yeast spp. with potential applications in industrial biotechnology. J Microbiol Biotechnol. 2016;26(11):1891–1907.
- Chen L, Li Y-P, Li X-X, et al. Isolation of 4,4'-bond secalonic acid D from the marine-derived fungus Penicillium oxalicum with inhibitory property against hepatocellular carcinoma. J Antibiot. 2019;72(1):34–44.
- Nguyen TH, Nguyen VD. Characterization and applications of marine microbial enzymes in biotechnology and probiotics for animal health. Adv Food Nutr Res. 2017;80:37–74.
- Rai M, Gade A, Zimowska B, et al. Marine-derived phoma – the gold mine of bioactive compounds. Appl Microbiol Biotechnol. 2018;102(21):9053–9066.
- Tang R, Kimishima A, Ishida R, et al. Selective cytotoxicity of epidithiodiketopiperazine DC1149B, produced by marine-derived Trichoderma lixii on the cancer cells adapted to glucose starvation. J Nat Med. 2020;74(1):153–158.
- Pang K-L, Overy DP, Jones EBG, et al. Marine fungi’ and ‘marine-derived fungi’ in natural product chemistry research: toward a new consensual definition. Fungal Biol Rev. 2016;30(4):163–175.
- Jones EBG, Suetrong S, Sakayaroj J, et al. Classification of marine ascomycota, basidiomycota, blastocladiomycota and chytridiomycota. Fungal Divers. 2015;73(1):1–72.
- Spatafora JW, Chang Y, Benny GL, et al. A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia. 2016;108(5):1028–1046.
- Gadanho M, Almeida JM, Sampaio JP. Assessment of yeast diversity in a marine environment in the South of Portugal by microsatellite-primed PCR. Antonie Van Leeuwenhoek. 2003;84(3):217–227.
- Chen YS, Yanagida F, Chen LY. Isolation of marine yeasts from coastal waters of northeastern Taiwan. Aquat Biol. 2009;8:55–60.
- Cathrine SJ, Raghukumar C. Anaerobic denitrification in fungi from the coastal marine sediments off Goa, India. Mycol Res. 2009;113(1):100–109.
- Gutiérrez MH, Pantoja S, Quiňones RA. First record of filamentous fungi in the coastal upwelling ecosystem off Central Chile. Gayana. 2010;74:66–73.
- Cury JC, Araujo FV, Coelho-Souza SA, et al. Microbial diversity of a Brazilian coastal region influenced by an upwelling system and anthropogenic activity. PLoS One. 2011;6(1):e16553.
- Arfi Y, Marchand C, Wartel M, et al. Fungal diversity in anoxic-sulfidic sediments in a mangrove soil. Fung Ecol. 2012;5(2):282–285.
- Li L, Singh P, Liu Y, et al. Diversity and biochemical features of culturable fungi from the coastal waters of Southern China. AMB Express. 2014;4:60.
- Wang Y, Sen B, He Y. Spatiotemporal distribution and assemblages of planktonic fungi in the coastal waters of the Bohai Sea. Front Microbiol. 2018;9:584.
- Richards TA, Leonard G, Mahé F, et al. Molecular diversity and distribution of marine fungi across 130 European environmental samples. Proc R Soc B. 2015;282(1819):20152243.
- Balabanova L, Slepchenko L, Son O, et al. Biotechnology potential of marine fungi degrading plant and algae polymeric substrates. Front Microbiol. 2018;9:1527.
- Borovec O, Vohnik M. Ontogenetic transition from specialized root hairs to specific root-fungus symbiosis in the dominant mediterranean seagrass Posidonia oceanica. Sci Rep. 2018;8(1):10773.
- Borzykh OG, Zvereva LV. Mycobiota of the giant oyster, Crassostrea gigas (Thunberg, 1787) (Bivalvia), from the Peter the Great Bay, Sea of Japan. Microbiology. 2012;81(1):109–111.
- Bovio E, Garzoli L, Poli A, et al. The culturable mycobiota associated with three Atlantic sponges, including two new species: Thelebolus balaustiformis and T. spongiae. Fungal Syst Evol. 2018;1:141–167.
- Gao Z, Johnson ZI, Wang G. Molecular characterization of the spatial diversity and novel lineages of mycoplankton in Hawaiian coastal waters. ISME J. 2010;4(1):111–120.
- Gao Z, Li BL, Zheng CC, et al. Molecular detection of fungal communities in the Hawaiian marine sponges Suberites zeteki and Mycale armata. Appl Environ Microbiol. 2008;74(19):6091–6101.
- King GM, Judd C, Kuske CR, et al. Analysis of stomach and gut microbiomes of the Eastern oyster (Crassostrea virginica) from coastal Louisiana, USA. PLoS One. 2012;7(12):e51475.
- Scholz B, Guillou L, Marano AV, et al. Zoosporic parasites infecting marine diatoms – a black box that needs to be opened. Fungal Ecol. 2016;19:59–76.
- Bass D, Howe A, Brown N, et al. Yeast forms dominate fungal diversity in the deep oceans. Proc Biol Sci. 2007;274(1629):3069–3077.
- Burgaud G, Hué NTM, Arzur D, et al. Effects of hydrostatic pressure on yeasts isolated from deep-sea hydrothermal vents. Res Microbiol. 2015;166(9):700–709.
- Damare S, Raghukumar C, Raghukumar S. Fungi in deep-sea sediments of the Central indian basin. Deep Sea Res Part I Oceanogr Res Pap. 2006;53(1):14–27.
- Lai X, Cao L, Tan H, et al. Fungal communities from methane hydrate-bearing deep-sea marine sediments in South China Sea. ISME J. 2007;1(8):756–762.
- Singh P, Raghukumar C, Verma P, et al. Phylogenetic diversity of culturable fungi from the deep-sea sediments of the Central Indian Basin and their growth characteristics. Fung Divers. 2010;40(1):89–102.
- Singh P, Raghukumar C, Meena RM, et al. Fungal diversity in deep-sea sediments revealed by culture-dependent and culture-independent approaches. Fungal Ecol. 2012;5(5):543–553.
- Nguyen AD, Zhao J-X, Feng Y-X, et al. Impact of recent coastal development and human activities on Nha Trang Bay, Vietnam: evidence from a Porites lutea geochemical record. Coral Reefs. 2013;32(1):181–193.
- Nguyen KAT, Nguyen TAT, Jolly C, et al. Economic efficiency of extensive and intensive shrimp production under conditions of disease and natural disaster risks in Khanh Hoa and Tra Vinh provinces, Vietnam. Sustainability. 2020;12(5):2140.
- Nguyen VD, Le MH, Trang ST. Application of probiotics from marine microbes for sustainable marine aquaculture development. In: Kim SK, editor. Marine microbiology: bioactive compounds and biotechnological applications. Weinheim: Wiley‐VCH; 2013. p. 307–349.
- Nguyen VD, Pham TT, Nguyen THT, et al. Screening of marine bacteria with bacteriocin-like activities and probiotic potential for ornate spiny lobster (Panulirus ornatus) juveniles. Fish Shellfish Immunol. 2014;40(1):49–60.
- Harju S, Fedosyuk H, Peterson KR. Rapid isolation of yeast genomic DNA: Bust n’ Grab. BMC Biotechnol. 2004;4:8.
- Toju H, Tanabe AS, Yamamoto S, et al. High-coverage ITS primers for the DNA-based identification of ascomycetes and basidiomycetes in environmental samples. PLoS One. 2012;7(7):e40863.
- White TJ, Bruns TD, Lee SB. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. New York: Academic Press; 1990. p. 315–322.
- Pham TT, Ho THN, Nguyen VD. Screening for bacteriocin-like antimicrobial activity against shrimp pathogenic vibrios and molecular identification of marine bacteria from otter clam Lutraria philippinarum. Thai J Vet Med. 2014;1:345–353.
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797.
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98.
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406–425.
- Kumar S, Stecher G, Li M, et al. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–1549.
- Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16(2):111–120.
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39(4):783–791.
- Magoc T, Salzberg S. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–2963.
- Li W, Fu L, Niu B, et al. Ultrafast clustering algorithms for metagenomic sequence analysis. Brief Bioinform. 2012;13(6):656–668.
- Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336.
- Nilsson RH, Larsson K-H, Taylor AFS, et al. The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019;47(D1):D259–D264.
- Ben Mefteh F, Frikha F, Daoud A, et al. Response surface methodology optimization of an acidic protease produced by Penicillium bilaiae isolate TDPEF30, a newly recovered endophytic fungus from healthy roots of date palm trees (Phoenix dactylifera L.). Microorganisms. 2019;7(3):74.
- Monga M, Goyal M, Kl K. Production and stabilization of amylases from Aspergillus niger. Mycosphere. 2011;2:129–134.
- Qasim SS, Shakir KA, Al-Shaibani AB. Isolation, screening and production of phytate degrading enzyme (phytase) from local fungal isolate. Iraqi J Agric Sci. 2016;47:121–128.
- Coniglio RO, Fonseca MI, Villalba LL, et al. Screening of new secretory cellulases from different supernatants of white rot fungi from Misiones, Argentina. Mycology. 2017;8(1):1–10.
- Abu-Tahon MA, Isaac GS. Anticancer and antifungal efficiencies of purified chitinase produced from Trichoderma viride under submerged fermentation. J Gen Appl Microbiol. 2020;66(1):32–40.
- Nguyen NH, Song Z, Bates ST, et al. FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016;20:241–248.
- Farr DF, Rossman AY. 2020. Fungal databases, U.S. National Fungus Collections, ARS, USDA [cited 2020 September 15]. Available from: https://nt.ars-grin.gov/fungaldatabases
- Smith ME, Douhan GW, Rizzo DM. Intra-specific and intra-sporocarp ITS variation of ectomycorrhizal fungi as assessed by rDNA sequencing of sporocarps and pooled ectomycorrhizal roots from a Quercus woodland. Mycorrhiza. 2007;18(1):15–22.
- Vu TD, Groenewald M, de Vries M, et al. Largescale generation and analysis of filamentous fungal DNA barcodes boosts coverage for kingdom Fungi and reveals thresholds for fungal species and higher taxon delimitation. Stud Mycol. 2019;92:135–154.
- Nguyen VD, Pham TT. 2021. Penicillium vietnamense sp. nov., the first novel marine fungi species described from Vietnam with a unique conidiophore structure and molecular phylogeny of Penicillium section Charlesia (submitted in parallel to Mycobiology).
- Nhi Cong LT, Ngoc Mai CT, Thanh VT, et al. Application of a biofilm formed by a mixture of yeasts isolated in Vietnam to degrade aromatic hydrocarbon polluted wastewater collected from petroleum storage. Water Sci Technol. 2014;70(2):329–336.
- Nguyen TTG, Nguyen TC, Leelakriangsak M. Promotion of Lactobacillus plantarum on growth and resistance against acute hepatopancreatic necrosis disease pathogens in white-leg shrimp (Litopenaeus vannamei). Thai J Vet Med. 2018;48:19–28.
- Quach TKN. Assessing the value of coral reefs in the face of climate change: the evidence from nha trang Bay. Vietnam. Ecosyst Serv. 2019;35:99–108.
- Kimura H, Naganuma T. Thraustochytrids: a neglected agent of the marine microbial food chain. Aquat Ecosyst Health Manag. 2001;4(1):13–18.
- Kimura H, Sato M, Sugiyama C, et al. Coupling of thraustochytrids and POM, and of bacterio- and phytoplankton in a semi-enclosed coastal area: implication for different substrate preference by the planktonic decomposers. Aquat Microb Ecol. 2001;25:293–300.
- Richards TA, Jones MDM, Leonard G, et al. Marine fungi: their ecology and molecular diversity. Ann Rev Mar Sci. 2012;4:495–522.
- Downs CA, Kramarsky-Winter E, Segal R, et al. Toxicopathological effects of the sunscreen UV filter, oxybenzone (benzophenone-3), on coral planulae and cultured primary cells and its environmental contamination in Hawaii and the U.S. Virgin in islands. Arch Environ Contam Toxicol. 2016;70(2):265–288.
- Comeau AM, Vincent WF, Bernier L, et al. Novel chytrid lineages dominate fungal sequences in diverse marine and freshwater habitats. Sci Rep. 2016;6:30120.
- Grossart H-P, Wurzbacher C, James TY, et al. Discovery of dark matter fungi in aquatic ecosystems demands a reappraisal of the phylogeny and ecology of zoosporic fungi. Fungal Ecol. 2016;19:28–38.
- Schoch CL, Seifert KA, Huhndorf S, et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi. Proc Natl Acad Sci USA. 2012;109(16):6241–6246.
- Ward DM, Weller R, Bateson MM. 16S rRNA sequences reveal numerous uncultured microorganisms in a natural community. Nature. 1990;345(6270):63–65.
- Sneath PH, Sokal RR. 1973. Numerical taxonomy. The principles and practice of numerical classification. San Francisco: Freeman WH.
- Taylor JW, Jacobson DJ, Kroken S, et al. Phylogenetic species recognition and species concepts in fungi. Fungal Genet Biol. 2000;31(1):21–32.
- Houbraken J, Kocsubé S, Visagie CM, et al. Classification of Aspergillus, Penicillium, Talaromyces and related genera (Eurotiales): an overview of families, genera, subgenera, sections, series and species. Stud Mycol. 2020;95:5–169.
- Barrett K, Jensen K, Meyer AS, et al. Fungal secretome profile categorization of CAZymes by function and family corresponds to fungal phylogeny and taxonomy: Example Aspergillus and penicillium. Sci Rep. 2020;10(1):5158.
- Danovaro R, Pusceddu A. Biodiversity and ecosystem functioning in coastal lagoons: does microbial diversity play any role? Estuar Coast Shelf Sci. 2007;75(1–2):4–12.
- Strom SL. Microbial ecology of ocean biogeochemistry: a community perspective. Science. 2008;320(5879):1043–1045.
- Damare SR, Nagarajan M, Raghukumar C. Spore germination of fungi belonging to Aspergillus species under deep-sea conditions. Deep Sea Res Part I Oceanogr Res Pap. 2008;55(5):670–678.
- Schauer F, Hanschke R. Taxonomy and ecology of the genus Candida. Mycoses. 1999;42(S1):12–21.
- Sandhu DK, Waraich MK. Yeasts associated with pollinating bees and flower nectar. Microb Ecol. 1985;11(1):51–58.
- Irinyi L, Serena C, Garcia-Hermoso D, et al. International Society of Human and Animal Mycology (ISHAM) – ITS reference DNA barcoding database-the quality controlled standard tool for routine identification of human and animal pathogenic fungi. Med Mycol. 2015;53(4):313–337.
- Buckley HR, van Uden N. Five new Candida species. Mycopathol Mycol Appl. 1968;36(3):257–266.
- Zaragoza S, Galanternik L, Vazquez M, et al. Candida blankii: new agent in cystic fibrosis airways? J Cyst Fibros. 2015;14:S140.
- Nobrega de Almeida J, Campos SV, Thomaz DY, et al. Candida blankii: an emergent opportunistic yeast with reduced susceptibility to antifungals. Emerg Microbes Infect. 2018;7(1):24.
- Benedict K, Richardson M, Vallabhaneni S, et al. Emerging issues, challenges, and changing epidemiology of fungal disease outbreaks. Lancet Infect Dis. 2017;17(12):e403–e411.
- Chowdhary A, Stielow JB, Upadhyaya G, et al. Candida blankii: an emerging yeast in an outbreak of fungaemia in neonates in Delhi, India. Clin Microbiol Infect. 2020;26(5):648.e5–648.e8.
- Magill SS, O’Leary E, Janelle SJ, et al. Changes in prevalence of health care-associated infections in U.S. hospitals. N Engl J Med. 2018;379(18):1732–1744.
- Chowdhary A, Sharma C, Meis JF. Candida auris: a rapidly emerging cause of hospital-acquired multidrug-resistant fungal infections globally. PLoS Pathog. 2017;13(5):e1006290.
- Teixeira MM, Moreno LF, Stielow BJ, et al. Exploring the genomic diversity of black yeasts and relatives (Chaetothyriales, Ascomycota). Stud Mycol. 2017;86:1–28.
- Zhu D, Ma Y, Wang G, et al. Identification of Candida tropicalis BH-6 and synergistic effect with Pantoea agglomerans BH-18 on hydrogen production in marine culture. Appl Biochem Biotechnol. 2015;175(5):2677–2688.
- Burgaud G, Arzur D, Sampaio JP, et al. Candida oceani sp. nov., a novel yeast isolated from a Mid-Atlantic Ridge hydrothermal vent (–2300 meters). Antonie Van Leeuwenhoek. 2011;100(1):75–82.
- Cong LTN, Ngoc Mai CT, Morikawa M, et al. Transformation of iso-pentylbenzene by a biofilm-forming strain of Candida viswanathii TH1 isolated from oil-polluted sediments collected in coastal zones in Vietnam. J Environ Sci Health A Tox Hazard Subst Environ Eng. 2014a;49(7):777–786.
- Bac ND, Anh LT, Quang LB, et al. Prevalence of Candida bloodstream isolates from patients in two hospitals in Vietnam. Iran J Microbiol. 2019;11(2):108–113.
- Thanh VN, Thuy NT, Chi NT, et al. New insight into microbial diversity and functions in traditional Vietnamese alcoholic fermentation. Int J Food Microbiol. 2016;232:15–21.
- León VM, García-Agüera I, Moltó V, et al. PAHs, pesticides, personal care products and plastic additives in plastic debris from Spanish Mediterranean beaches. Sci Total Environ. 2019;670:672–684.
- Naranjo-Ortiz MA, Gabaldón T. Fungal evolution: major ecological adaptations and evolutionary transitions. Biol Rev Camb Philos Soc. 2019;94(4):1443–1476.
- Bonugli-Santos RC, Dos Santos Vasconcelos MR, Passarini MR. Marine-derived fungi: diversity of enzymes and biotechnological applications. Front Microbiol. 2015;10:6:269.
- Cunliffe M, Hollingsworth A, Bain C, et al. Algal polysaccharide utilisation by saprotrophic planktonic marine fungi. Fungal Ecol. 2017;30:135–138.
- McGenity TJ, Folwell BD, McKew BA, et al. Marine crude-oil biodegradation: a central role for interspecies interactions. Aquat Biosyst. 2012;8(1):10.
- Passarini MRZ, Rodrigues MVN, da Silva M, et al. Marine-derived filamentous fungi and their potential application for polycyclic aromatic hydrocarbon bioremediation. Mar Pollut Bull. 2011;62(2):364–370.
- Steliga T, Jakubowicz P, Kapusta P. Changes in toxicity during in situ bioremediation of weathered drill wastes contaminated with petroleum hydrocarbons. Bioresour Technol. 2012;125:1–10.
- Uribe-Alvarez C, Ayala M, Perezgasga L, et al. First evidence of mineralization of petroleum asphaltenes by a strain of Neosartorya fischeri. Microb Biotechnol. 2011;4(5):663–672.
- Sanders R, Henson SA, Koski M, et al. The biological carbon pump in the North atlantic. Prog Oceanogr. 2014;129:200–218.
- Morán XAG, Gasol JM, Pernice MC, et al. Temperature regulation of marine heterotrophic prokaryotes increases latitudinally as a breach between bottom-up and top-down controls. Glob Change Biol. 2017;23(9):3956–3964.