
Abstract
The identification of oleaginous yeast species capable of simultaneously utilizing xylose and glucose as substrates to generate value-added biological products is an area of key economic interest. We have previously demonstrated that the Cutaneotrichosporon dermatis NICC30027 yeast strain is capable of simultaneously assimilating both xylose and glucose, resulting in considerable lipid accumulation. However, as no high-quality genome sequencing data or associated annotations for this strain are available at present, it remains challenging to study the metabolic mechanisms underlying this phenotype. Herein, we report a 39,305,439 bp draft genome assembly for C. dermatis NICC30027 comprised of 37 scaffolds, with 60.15% GC content. Within this genome, we identified 524 tRNAs, 142 sRNAs, 53 miRNAs, 28 snRNAs, and eight rRNA clusters. Moreover, repeat sequences totaling 1,032,129 bp in length were identified (2.63% of the genome), as were 14,238 unigenes that were 1,789.35 bp in length on average (64.82% of the genome). The NCBI non-redundant protein sequences (NR) database was employed to successfully annotate 11,795 of these unigenes, while 3,621 and 11,902 were annotated with the Swiss-Prot and TrEMBL databases, respectively. Unigenes were additionally subjected to pathway enrichment analyses using the Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Cluster of Orthologous Groups of proteins (COG), Clusters of orthologous groups for eukaryotic complete genomes (KOG), and Non-supervised Orthologous Groups (eggNOG) databases. Together, these results provide a foundation for future studies aimed at clarifying the mechanistic basis for the ability of C. dermatis NICC30027 to simultaneously utilize glucose and xylose to synthesize lipids.
1. Introduction
The use of lignocellulose as a biofuel holds great economic promise, as it is readily produced through secondary growth by woody plants and is composed of lignin, cellulose, and hemicellulose [Citation1,Citation2]. When pretreated appropriately, lignocellulose-containing biomass can yield substantial quantities of xylose and glucose, as well as other sugar byproducts [Citation3,Citation4]. The development of novel approaches to processing lignocellulose hydrolysate in an efficient manner to generate value-added biological products would thus be beneficial from both an economic and an ecological perspective [Citation5,Citation6].
The efficient utilization of lignocellulose necessitates the assimilation of both xylose and glucose, given that these are its two main hydrolytic byproducts [Citation7]. However, most microbes are only able to use glucose as an energy source, and those that can utilize xylose generally suppress its metabolic processing when glucose is available through a process known as carbon catabolite repression [Citation8–10]. Such repression enables microbes to preferentially metabolize carbon sources that are more easily processed by suppressing the expression and activity of enzymes associated with the processing of non-preferred carbon sources [Citation11,Citation12]. The identification of oleaginous yeasts capable of simultaneously utilizing both xylose and glucose has been an active area of research interest, with several such yeast strains having been reported to date including Trichosporon cutaneum (present name, Cutaneotrichosporon cutaneum) and Cystobasidium iriomotense [Citation13,Citation14].
In a prior investigation, our team obtained a yeast strain capable of producing lipids from the soil in Nanyang, China. This strain was identified as Cutaneotrichosporon dermatis using morphological and phylogenetic analysis and is now housed in the Nanyang Center of Industrial Culture Collection (NICC, Henan Province, China; S.No: NICC30027) [Citation15]. Regardless of the glucose/xylose ratios, strain NICC30027 could efficiently absorb glucose and xylose simultaneously to accumulate a significant quantity of lipid when mixed sugars were employed as the carbon source. The biomass and lipid concentrations were maximum when the glucose/xylose ratio was 2:1, attaining 19.85 ± 0.39 and 9.53 ± 0.60 g/L, respectively [Citation15]. Additionally, strain NICC30027 largely produced fatty acids in the C16 and C18 series with content comparable to that of vegetable oil, suggesting that this microorganism may be well suited for use in biodiesel production [Citation15].
Cutaneotrichosporon dermatis is ubiquitous and was originally isolated from subaerial zones [Citation16]. This species was moved from the Trichosporon genus to Cutaneotrichosporon by Liu et al. [Citation17]. Due to its ability to manufacture several essential enzymes associated with human tissues, C. dermatis has been described as an opportunistic pathogen responsible for infections of the skin, blood, and nails [Citation18]. In addition, C. dermatis has resistance to some antifungal agents such as echinocandins and fluconazole [Citation19].
However, as genomic information pertaining to this yeast strain are not currently available, the molecular mechanisms whereby it is able to simultaneously metabolize both glucose and xylose to generate lipids remain unclear. In addition, without genomic sequence information of the strain NICC30027, efforts to conduct the precise metabolic engineering thereof remain challenging.
Herein, we present a draft genome assembly for C. dermatis NICC30027 in an effort to identify genes responsible for microbial lipid accumulation in this species and to clarify the molecular mechanisms governing simultaneous glucose and xylose utilization therein.
2. Materials and methods
2.1. Microbial culture
The NICC30027 strain was cultured in YPD medium (glucose 20 g/L, yeast extract 10 g/L, and peptone 10 g/L) at 30°C with constant shaking (180 rpm) [Citation15]. Cultures were established by inoculating a single colony in 10 mL of YPD medium in a 50 mL flask and culturing the cells overnight, followed by the transfer of 1 mL of this solution to 100 mL of YPD medium in a 500 mL flask. After an additional 72 h culture period, cells were centrifuged, snap-frozen with liquid nitrogen, and stored at −80°C.
2.2. Genomic DNA extraction, library construction and sequencing
Total gDNA was prepared with a Magnetic Plant Genomic DNA Kit (Cat. No. DP342-T1, Tiangen Biotech, China) based on provided directions [Citation20,Citation21], after which the concentration and purity of the resultant DNA were assessed using a Qubit fluorometer and a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific, CA, USA) [Citation22,Citation23], while 0.5% agarose gel electrophoresis was used to assess DNA integrity [Citation24,Citation25].
A DNBSEQ-2000 platform and a PacBio Sequel I system was used to conduct DNA sequencing at Beijing Genomics Institute (BGI, Shenzhen, China). Each sample sequencing library was constructed separately, and a specific barcode sequence was included in the adapter for the separation of pooling sequencing data. A DNBSEQ sequencing library was prepared with a 350 bp insert size for 150 bp paired-end sequencing [Citation26]. Briefly, a g-TUBE instrument (Covaris, USA) was used to fragment 1 μg of prepared gDNA based on provided directions, after which magnetic beads were used to select fragments from 200 to 400 bp in size, on average. The resultant fragments were then subjected to end-repair, 3′ adenylation, and adapter ligation. Magnetic beads were utilized to purify all PCR products. Resultant dsDNA PCR products were subjected to heat denaturation, after which the splint oligo sequence was used for circularization to yield a sing-stranded circular DNA (ssCirDNA) library that was subjected to quality control analyses. For PacBio sequencing, the insert size was instead >10 Kb. For this sequencing approach, a g-TUBE instrument was used to shear 1 μg of gDNA into fragments 10–15 Kb in size, after which a SMRTbell® Express Template Preparation Kit 2.0 (Pacific Biosciences, USA) was used for library construction. Briefly, these fragments were processed to remove single-stranded overhangs, DNA damage was repaired, and then end-repair, adenylation, and barcoded overhanging adapter ligation were performed. AMPure® PB Beads were used to purify all samples, which were then pooled, and fragments <10 Kb in size were removed with a BluePippin Size selection system (Sage Science, USA). A Qubit DNA HS Assay Kit and a Qubit fluorometer (Thermo Fisher Scientific) were used to quantify library DNA concentrations, while library sizing was assessed with an Agilent 2100 Bioanalyzer Instrument and an HS DNA Kit (Agilent Technologies, CA, USA). Following the annealing of the sequencing primer v4 to the SMRTbell template, the complex was bound by DNA polymerase (Sequel II Binding Kit 2.0, Pacific Biosciences, USA). AMPure PB Bead Purification was then used to remove free primers and polymerase, after which a Sequel Sequencing Kit 2.0 (PacBio) was used for library sequencing, with 10 h videos being captured for each SMRT Cell 8 M with the Sequel II sequencing platform (BGI-Shenzhen, China) [Citation27].
2.3. Genome assembly
The assembly of raw sequencing data from the DNBSEQ-2000 platform was conducted as follows: (1) Reads harboring adapter sequences were removed; (2) reads with a low-quality base ratio <50% (base quality ≤12) were removed; (3) reads with >10% unknown (‘N’) bases were removed. The remaining cleaned high-quality filtered data were then used for downstream bioinformatics analyses. Raw PacBio RSII sequencing data were processed as follows: (1) reads <1,000 bp in length were removed; (2) reads with a quality score <0.8 were removed; (3) adapter sequences were removed, with subreads being produced and subjected to the same length and quality cutoff criteria as defined above. These subreads were then corrected with Proovread 2.12 (-t 4 –coverage 60 –mode sr) to generate credible Corrected Reads. Celera Assembler 8.3 (doTrim_initialQualityBased = 1, doTrim_finalEvidenceBased = 1, doRemoveSpurReads = 1, doRemoveChimericReads = 1, and -d properties -U) and Falcon v0.3.0 (-v -dal8 -t32 -h60 -e.96 -l500 -s100 -H3000) were used to assemble corrected reads. GATK v1.6–13 was then utilized to correct single bases from the assembly results with the following parameters: -cluster 2 -window 5 -stand_call_conf 50 -stand_emit_conf 10.0 -dcov 200 MQ0 ≥ 4. SSPACE_Basic_v2.0 was utilized for scaffold construction, and gap-filling was performed with pbjelly2 15.8.24 (default parameters) [Citation28].
2.4. Gene annotation
Genewise 2.20, SNAP v 2010-07-28, Augustus 3.2.1, and GeneMarkes 4.21 were used to predict genes, while RNAmmer (v 1.2), tRNAs can-SE (v 1.3.1), and Rfam (v 9.1) were respectively used to identify rRNA, tRNA, and sRNA sequences. The transposon Repbase database, Repeat Protein Masker software, and De novo approaches were used for transposon analyses of the sequencing results. Tandem Repeat Finder (TRF v 4.04) was used for tandem repeat sequence identification.
Databases used for the identification, annotation, and functional analysis of identified protein-coding genes included the Gene Ontology (GO) (releases_2019-07-01), Kyoto Encyclopedia of Genes and Genomes (KEGG) (v 76), Cluster of Orthologous Groups of proteins (COG) (v 2014-11-10), Swiss-Prot (v 2016-01), TrEMBL (v 2016-01), NR (v 2015-05-31), evolutionary genealogy of genes: Non-supervised Orthologous Groups (eggNOG) (v 4.5), Antibiotic Resistance Genes Database (ARDB) (v 1.1), Virulence Factor Database (VFDB) (v 2019-07), Transporter Classification Database (TCDB) (v 2.0), Antibiotic Resistance Gene-ANNOTation (ARG-ANNOT) (v V6), Carbohydrate-active enzymes (CAZymes) (v 2016-04), Fungal Cytochrome P450 Database (FCPD) (v 1.1), Fungal Transcription Factor Database (FTFD), Comprehensive Antibiotic Resistance Database (CARD), Cell Wall Degrading Enzyme (CWDE), and Clusters of orthologous groups for eukaryotic complete genomes (KOG) databases, all of which were used with the default parameters.
3. Results
3.1. Genome features
Our sequencing analyses led to the generation of a 39,305,439 bp C. dermatis NICC30027 draft genome with a 60.15% GC content containing 37 scaffolds with an N50 of 2,902,125 bp ( and ). This genome was predicted to encode 14,238 protein-coding genes that were an average of 1,789.35 bp in length (64.82% of the genome) (Table S1). Predictive analyses additionally led to the identification of non-coding RNAs (ncRNAs) including 524 tRNAs, 142 sRNAs, 53 miRNAs, 28 snRNAs, and eight rRNA clusters (). tRNAs associated with all 20 amino acids were identified (Table S2). 1,032,129 bp of repeat sequences (2.63% of the genome) were identified. Transposon analyses additionally identified 268,220 bp of Long Terminal Repeat (LTR), 81,092 bp of Long Interspersed Elements (LINE), and 16,953 bp of Short Interspersed Elements (SINE) sequences ().
Figure 1. Circular Cutaneotrichosporon dermatis NICC30027 genome map. The Circos software was used to generate a genome map with six total circles. From outermost to innermost, the circles represent the following: 1–37 scaffolds sorted by length, with each arcuate column corresponding to a scaffold; 2 – gene density as determined by the number of genes in non-overlapping 50,000 bp windows, with greater density being indicated by darker coloration; 3 – ncRNA density as determined by the number of ncRNAs in non-overlapping 100,000 bp windows, with darker colors being indicative of lower ncRNA density values; 4 – repeat coverage in non-overlapping 50,000 bp windows, with greater coverage being denoted by darker coloration; 5 – GC content in non-overlapping 20,000 bp windows; 6 – GC skew [(G + C)/(G − C)] in non-overlapping 20,000 bp windows, with peaks inside/outside the circle indicating values lower than and higher than 1, respectively.
![Figure 1. Circular Cutaneotrichosporon dermatis NICC30027 genome map. The Circos software was used to generate a genome map with six total circles. From outermost to innermost, the circles represent the following: 1–37 scaffolds sorted by length, with each arcuate column corresponding to a scaffold; 2 – gene density as determined by the number of genes in non-overlapping 50,000 bp windows, with greater density being indicated by darker coloration; 3 – ncRNA density as determined by the number of ncRNAs in non-overlapping 100,000 bp windows, with darker colors being indicative of lower ncRNA density values; 4 – repeat coverage in non-overlapping 50,000 bp windows, with greater coverage being denoted by darker coloration; 5 – GC content in non-overlapping 20,000 bp windows; 6 – GC skew [(G + C)/(G − C)] in non-overlapping 20,000 bp windows, with peaks inside/outside the circle indicating values lower than and higher than 1, respectively.](/cms/asset/f65a90cd-44c1-4acb-8a2f-e10112085b8e/tmyb_a_2038844_f0001_c.jpg)
Table 1. General genome features of Cutaneotrichosporon dermatis NICC30027.
3.2. Gene annotation
Of the identified unigenes, 11,795 matched known genes in the NR database, while 3,621 unigenes yielded optimal hits in the Swiss-Prot database, and 11,902 matched known genes in the TrEMBL database ( and Table S3–S5).
Table 2. The results of gene annotation in Cutaneotrichosporon dermatis NICC30027.
A total of 70 candidate virulence factor genes were identified using the VFPB database (Table S6). While the CARD, ARG-ANNOT, and ARDB were employed to identify putative antimicrobial resistance genes, no such candidate genes were detected. A total of 36 genes encoding plant cell wall degradation-related enzymes were annotated with the CWDE database, while the FTFD and FCPD databases were respectively used to annotate 407 transcription factor-encoding and cytochrome P450-encoding genes ( and Table S7–S9).
Carbohydrate metabolism-associated enzymes were annotated with the CAZy database (, S10). In total, this approach identified 318 enzymes that were grouped into five categories including 34 proteins related to auxiliary activities (AAS), 56 related to carbohydrate-binding modules (CBMS), 129 glycoside hydrolases (GHS), 90 glycosyltransferases (GTS), and 9 polysaccharide lyases (PLS). Protein kinase and phosphatase predictions were made using the EKPD database. The 175 putative protein kinases identified via this approach included 23 AGC kinases, 20 atypical kinases, 26 calcium/calmodulin-dependent kinases (CAMK), 13 creatine kinase (CK) enzymes, 34 CMGC members, 23 sterile (STE) kinases, 2 tyrosine kinase-like (TKL) proteins, and 44 other proteins (Table S11). In addition, this approach identified 57 putative protein phosphatases (Table S12).
Table 3. The results of gene annotation using the databases CAZy and EKPD.
The TCDB database identified 608 transporter coding genes that were grouped into 16 families, of which porters (uniporters, symporters, antiporters), P-P-bond-hydrolysis-driven, transporters, oxidoreduction-driven transporters, were the most abundant, respectively containing 202, 143, and 60 unigenes (Table S13).
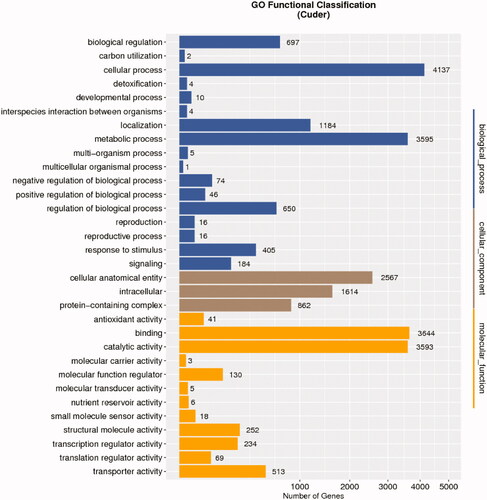
Blast2GO (http://www.BLAST2go.com) was used to conduct a GO term enrichment analysis revealing 11,030, 8,508, and 5,043 genes to be associated with defined biological process (BP), molecular function (MF), and cellular component (CC) annotations, respectively. Top enriched BP terms included “metabolic process,” “cellular process,” “single-organism process,” and “localization and biological regulation,” while top MF terms included “binding,” “catalytic activity,” and “transporter activity,” and top CC terms included “membrane,” “cell,” and “cell part” (, and Table S14, S15).
Figure 2. GO classification of all unigenes in Cutaneotrichosporon dermatis NICC30027.
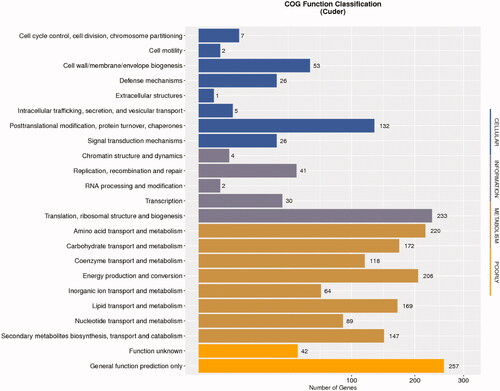
Gene annotation analyses revealed that of the 14,238 putative protein-coding genes identified in this study, 1,738 could be classified into COG families across 23 functional categories, with the most abundant terms being: “general function prediction only,” “translation, ribosomal structure and biogenesis,” and “amino acid transport and metabolism” ( and Table S16).
Figure 3. COG classification of all unigenes in Cutaneotrichosporon dermatis NICC30027.
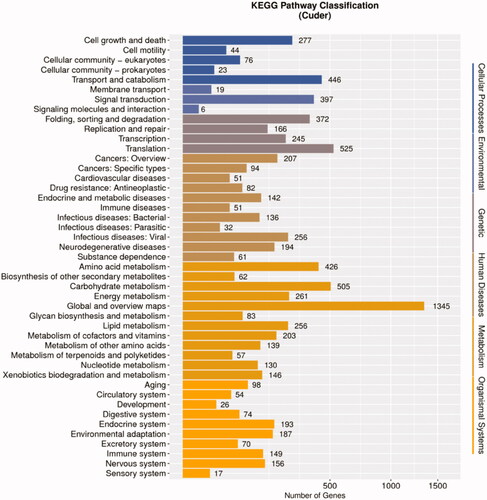
A KEGG pathway enrichment approach successfully classified 5554 genes into 376 pathways ( and Table S17), including 19 metabolic pathways containing over 100 unigenes. Metabolic pathways (ko:01100) was the top enriched KEGG term, containing 1241 unigenes, followed by the biosynthesis of secondary metabolites (ko: 01110), biosynthesis of antibiotics (ko: 01130), microbial metabolism in diverse environments (ko: 01120), and ribosome (ko: 03010) pathways, which respectively contained 526, 390, 359, and 184 unigenes.
Figure 4. KEGG classification of all unigenes in Cutaneotrichosporon dermatis NICC30027.
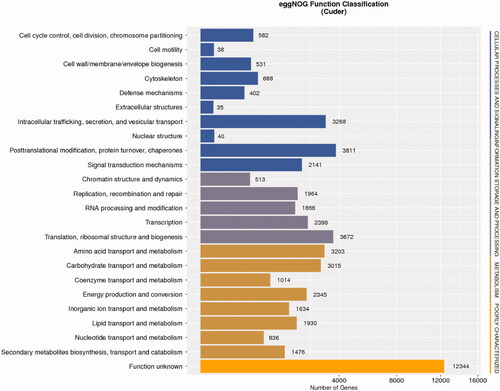
Unigene alignment to the eggNOG database was additionally performed to classify their predicted functions, ultimately grouping 9,163 unigenes into 24 categories ( and Table S18). The “function unknown” NOG category was the largest such grouping, followed by the “posttranslational modification, protein turnover, chaperones,” “translation, ribosomal structure and biogenesis,” “intracellular trafficking, secretion, and vesicular transport,” and “amino acid transport and metabolism” categories, whereas the “extracellular structures” category was the smallest.
Figure 5. eggNOG classification of all unigenes in Cutaneotrichosporon dermatis NICC30027.
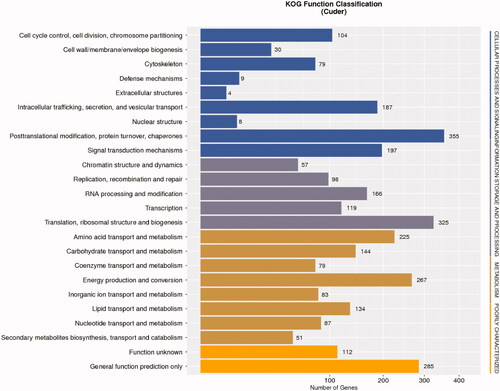
The KOG database was additionally leveraged for functional prediction analyses of these unigenes, leading to the subcategorization of 2,884 unigenes into 24 KOG classes ( and Table S19). Of these, the “posttranslational modification, protein turnover, chaperones,” “translation, ribosomal structure, and biogenesis,” and “translation, ribosomal structure, and biogenesis” categories were the largest.
Figure 6. KOG classification of all unigenes in Cutaneotrichosporon dermatis NICC30027.
3.3. Nucleotide sequence accession numbers
The complete C. dermatis NICC30027 draft genome sequence has been deposited at DDBJ/ENA/GenBank (accession number JAIGNX000000000.1). The version described in this paper is the first version, JAIGNX000000000.1.
4. Discussion
Genome sequencing has become an increasingly commonplace and straightforward molecular biology technique [Citation29]. Different sequencing platforms use different methods to construct sequencing libraries, which may cause differences in the subsequent analysis processes [Citation30,Citation31]. In this study, a DNBSEQ-2000 platform and a PacBio Sequel I system were used to conduct DNA sequencing at BGI, which is among the biggest genomic research institutions in the world, with several sequencing systems [Citation30]. To date, sequencing has been performed for the genomes of 8 species and 13 strains in the Cutaneotrichosporon genus ().
Table 4. The strains of the genus Cutaneotrichosporon whose genomes have been sequenced.
Of these, the C. mucoides JCM 9939 strain exhibits the largest genome (∼40.8 Mb), while C. curvatum SBUG-Y 855 exhibits the smallest (∼16.4 Mb). The published genome sizes of different strains of the same species also differ markedly, as is the case for C. cutaneum ACCC 20271 (∼30.4 Mb) and C. cutaneum JCM 1462 (∼23.2 Mb). A similar phenomenon has also been observed among C. dermatis strains, with the C. dermatis NICC30027 genome sequenced in this study being 39,305,439 bp long, whereas the genome of C. dermatis JCM 11170 submitted by Takashima et al. [Citation32] was 23,337,637 bp in length – a difference of 15,967,802 bp. Genomic size differences among these strains may be attributable to strain-specific differences or to the distinct sequencing technologies used in these prior studies.
Genes encoding xylose reductase, xylitol dehydrogenase, and xylulo kinase were identified within the NICC30027 genome (). As such, we hypothesize that NICC30027 metabolize xylose via the following mechanism: NADPH-dependent xylose reductase initially reduces xylose to xylitol, after which NAD-dependent xylitol dehydrogenase oxidizes xylitol to xylulose, which is then phosphorylated by xylulo kinase to yield xylose-5-phosphate, which subsequently enters into the pentose phosphate pathway (PPP).
Table 5. Some key enzymes in xylose utilization and lipid synthesis pathways.
The sugar transport system is critical for the absorption and utilization of sugar, which differs between species and substrates [Citation36,Citation37]. Numerous xylose transporters have been found in yeasts of various species [Citation37]. In general, xylose can be transported either through hijacking the glucose transporters or through specific transporter [Citation37]. However, most glucose transporters have a higher affinity for glucose than xylose [Citation38]. As a result of competitive inhibition during the co-fermentation of glucose and xylose, xylose transport is inhibited in the presence of glucose [Citation38]. In xylose-fermenting yeast such as Candida shehatae, Scheffersomyces stipitis, Candida intermedia, and Kluyveromyces marxianus, xylose specific transporters were discovered [Citation39–41]. To absorb xylose, the majority of xylose-specific transporters participate in the proton symport system [Citation37]. Due to the high affinity of the proton symport system for xylose and the fact that it does not interfere with glucose transport, xylose is often carried very effectively via the proton symport system [Citation37]. Few xylose-fermenting yeast species, such as C. intermedia and S. stipitis, have been found to contain this transport mechanism [Citation37,Citation41]. The gene encoding xylose/protocol symporter (GME7379_g) was identified within the NICC30027 genome (). This implies that C. dermatis NICC30027 assimilates the xylose through the proton symptom mechanism, which ensures the effective absorption of xylose in the presence of glucose. That is to say, xylose/protocol symporter is an important guarantee for C. dermatis NICC30027 to utilize glucose and xylose simultaneously.
In many microbial cells, xylose metabolism is inhibited by glucose due to carbon catabolite repression (CCR), and the catabolite activator protein (CAP) and cAMP complex are necessary for the expressions of key genes involved in xylose metabolism [Citation42,Citation43]. Unfortunately, it is difficult to entirely reveal how CCR functions due to the complex nature of CCR [Citation44,Citation45]. Several strategies such as adaptive evolution, rational design and computation simulation, have been conducted to relax CCR by different researchers [Citation43,Citation46]. Yet, the modified strains created by these techniques are incapable of efficiently co-utilizing xylose and glucose. The strain C. dermatis NICC30027 demonstrated natural capacity to consume xylose and glucose [Citation15]. In our opinion, one of the following conditions exists in the strain C. dermatis NICC30027: i) there is a high level of the cAMP-CAP complex in the cell due to some unknown metabolic mechanisms; ii) the expressions of key genes involved in xylose metabolism do not require cAMP-CAP mediated activation. Objectively, the molecular mechanisms governing simultaneous glucose and xylose utilization in C. dermatis NICC30027 is complex, and it is difficult to entirely reveal this mechanism only by genome sequencing and gene annotation. In the future, we will further analyze the mechanisms through transcriptome sequencing, proteome analysis, and development of the metabolic model and so on.
NADPH is essential for the synthesis of fatty acids, and is primarily produced by the malic enzyme (ME), glucose-6-phosphate dehydrogenase (ZWF1), NADP-dependent isocitric dehydrogenase (IDP1), and 6-phosphogluconate dehydrogenase (GND) [Citation47,Citation48]. We were able to identify genes predicted to encode all four of these enzymes within the NICC30027 genome (). In the context of de novo fatty acid synthesis, acetyl-CoA serves as a substrate that is generated via ATP-citrate lyase (ACL)-mediated citrate cleavage, after which it is converted to yield malonyl-CoA by acetyl-CoA carboxylase (ACC) [Citation49]. We successfully identified both ACL and ACC genes within the C. dermatis NICC30027. Fatty acid synthase (FAS) activity in C. dermatis NICC30027 was predicted to be mediated by the Fas1 and Fas2 genes (beta and alpha subunits, respectively) ().
Prior reports have identified C. dermatis as a potentially infectious yeast species that has been detected in the blood, skin, and nails [Citation18,Citation50]. We identified 70 putative virulence factor genes within the C. dermatis NICC30027 genome. Based on our tentative findings and literature support, we speculate that C. dermatis may achieve pathogenicity owing to its ability to produce enzymes that are damaging to human tissues, including urease, esterase, and lipase. While some studies have reported that certain C. dermatis isolates may be resistant to several common clinical antifungal agents including echinocandins and amphotericin B, we detected no evidence of antifungal resistance genes within the genome of strain NICC30027 [Citation18,Citation19]. As such, whether it is able to resist antifungal treatment warrants further study.
With the increasing amount of genetic data being posted in public databases, an increasing number of gene annotation methods have been created [Citation51]. These gene annotation approaches make it possible to analyze massive amounts of newly sequenced data. They may, however, result in incorrect functional annotations [Citation52]. Annotating genomes correctly is critical for our knowledge and use of functional gene diversity [Citation53,Citation54]. In order to confirm the accuracy of the gene annotations, structural analysis or assessment of expression level is usually necessary [Citation55]. As the function of a protein is determined by its structure, structural analysis is an important method for predicting protein function and confirming the accuracy of the gene annotations [Citation55]. The structural characteristics of proteins include overall fold, surface clefts and binding pockets, active sites and so on [Citation56]. Frequently, proteins with comparable activities have identical structures. Thus, identifying a structural resemblance between the target protein and known-function proteins may provide insight into possible function types [Citation55]. Several methods can be used for fold searching, for example, Dali Domain Dictionary (DALI), Secondary Structure Matching (SSM) and Graph-theoretic program (GRATH) [Citation57–59]. Due to the fact that substrates and regulatory regions typically attach in cavities on the surface, examination of the clefts and binding pockets can reveal a wealth of information valuable for predicting protein activity [Citation55]. The web server Pocket and Void Surfaces Of Amino acid Residues (PvSOAR) and the program SURFNET proposed by Laskowski can be used to analyze protein clefts and binding pockets [Citation60–62]. The active sites of proteins are strongly conserved over evolutionary time [Citation55]. Predictions of protein functions based on active sites can be conducted using the conserved functional group (CFG) analysis and evolutionary trace (ET) [Citation63,Citation64]. Numerous methods for measuring the quantities of mRNA molecules and proteins can be utilized to determine the expression level of anticipated genes [Citation65]. Reverse transcription-polymerase chain reaction (RT-PCR), northern blotting, microarrays, and transcriptome sequencing are all common methods for determining mRNA levels [Citation66]. Protein levels can be determined using enzyme-linked immunosorbent assays (ELISA) and western blotting, as well as protein microarrays and proteomic analysis [Citation67,Citation68].
In the future, we will use the above analysis methods and databases to analyze the protein structure of predicted genes. Furthermore, we will confirm the accuracy of gene annotation through experiments. Transcriptomes of the strain C. dermatis NICC30027 under different culture conditions will be sequenced to assess the expression levels and compare the differentially expressed genes. The interesting genes will be verified by RT-PCR. In addition, protein chip technology will be used to analyze the expression level of proteins. Western blotting will be used to verify the expression of some specific genes.
In conclusion, we successfully assembled a draft version of the C. dermatis NICC30027 genome. We further identified genes encoding proteins and ncRNAs within this whole-genome sequence and employed several databases to tentatively annotate and classify the protein-coding genes detected within this yeast strain. Notably, we identified multiple genes associated with xylose metabolism and lipid synthesis, and we analyzed the xylose metabolic pathway in detail. Together, our findings offer a robust foundation for future analytical and bioengineering efforts aimed at understanding and enhancing the simultaneous processing of glucose and xylose by C. dermatis NICC30027 to yield lipids suitable for biodiesel production or other applications.
Author contributions
S.G. and L.W. conceived and designed the study. L.W., Z.B., S.W., Y.C., B.L. and S.C. performed the experiments. L.W., S.G., and C.W. performed all data analysis. L.W., Y.W., and Q.M. wrote and revised the manuscript. All authors read and approved the final manuscript.
Supplemental Material
Download MS Word (11.9 KB)Supplemental Material
Download Zip (11.4 MB)Disclosure statement
The authors declare no competing financial interest.
Data availability statement
All data are provided in full in the results section of this paper apart from the genome sequence of Cutaneotrichosporon dermatis NICC30027 which is available at www.ncbi.nlm.nih.gov/nuccore/ under accession number JAIGNX000000000.1.
Additional information
Funding
References
- Roy S, Dikshit PK, Sherpa KC, et al. Recent nanobiotechnological advancements in lignocellulosic biomass valorization: a review. J Environ Manage. 2021;297:113422.
- Kumar D, Singh B, Korstad J, et al. Utilization of lignocellulosic biomass by oleaginous yeast and bacteria for production of biodiesel and renewable diesel. Renewable Sustainable Energy Rev. 2017;73:654–671.
- Akhlisah ZN, Yunus R, Abidin ZZ, et al. Pretreatment methods for an effective conversion of oil palm biomass into sugars and high-value chemicals. Biomass Bioenergy. 2021;144:105901.
- Abdel-Rahman MA, Tashiro Y, Sonomoto K, et al. Lactic acid production from lignocellulose-derived sugars using lactic acid bacteria: overview and limits. J Biotechnol. 2011;156(4):286–301.
- Hoang AT, Nižetić S, Ong HC, et al. Insight into the recent advances of microwave pretreatment technologies for the conversion of lignocellulosic biomass into sustainable biofuel. Chemosphere. 2021;281:130878.
- Awasthi MK, Sarsaiya S, Patel A, et al. Refining biomass residues for sustainable energy and bio-products: an assessment of technology, its importance, and strategic applications in circular bio-economy. Renewable Sustainable Energy Rev. 2020;127:109876.
- Bhatia SK, Kim S-H, Yoon J-J, et al. Current status and strategies for second generation biofuel production using microbial systems. Energy Convers Manage. 2017;148:1142–1156.
- Tanimura A, Takashima M, Sugita T, et al. Lipid production through simultaneous utilization of glucose, xylose, and L-arabinose by Pseudozyma hubeiensis: a comparative screening study. AMB Express. 2016;6(1):58.
- Brandenburg J, Blomqvist J, Pickova J, et al. Lipid production from hemicellulose with Lipomyces starkeyi in a pH regulated fed-batch cultivation. Yeast. 2016;33(8):451–462.
- Tanadul O-U-M, Noochanong W, Jirakranwong P, et al. EMS-induced mutation followed by quizalofop-screening increased lipid productivity in chlorella sp. Bioprocess Biosyst Eng. 2018;41(5):613–619.
- Sarkar P, Goswami G, Mukherjee M, et al. Heterologous expression of xylose specific transporter improves xylose utilization by recombinant Zymomonas mobilis strain in presence of glucose. Process Biochem. 2021;102:190–198.
- Sun T, Yu Y, Wang K, et al. Engineering Yarrowia lipolytica to produce fuels and chemicals from xylose: a review. Bioresour Technol. 2021;337:125484.
- Hu C, Wu S, Wang Q, et al. Simultaneous utilization of glucose and xylose for lipid production by Trichosporon cutaneum. Biotechnol Biofuels. 2011;4(1):25.,
- Tanimura A, Sugita T, Endoh R, et al. Lipid production via simultaneous conversion of glucose and xylose by a novel yeast, Cystobasidium iriomotense. PLOS One. 2018;13(9):e0202164.
- Wang L, Wang D, Zhang Z, et al. Comparative glucose and xylose coutilization efficiencies of soil-isolated yeast strains identify Cutaneotrichosporon dermatis as a potential producer of lipid. ACS Omega. 2020;5(37):23596–23603.
- Gadanho M, Sampaio JP. Occurrence and diversity of yeasts in the mid-atlantic ridge hydrothermal fields near the Azores Archipelago. Microb Ecol. 2005;50(3):408–417.
- Liu X-Z, Wang Q-M, Göker M, et al. Towards an integrated phylogenetic classification of the tremellomycetes. Stud Mycol. 2015;81:85–147.
- do Espírito Santo EPT, Monteiro RC, da Costa ARF, et al. Molecular identification, genotyping, phenotyping, and antifungal susceptibilities of medically important Trichosporon, Apiotrichum, and Cutaneotrichosporon species. Mycopathologia. 2019;185:307–317.
- Pagani DM, Heidrich D, Paulino GVB, et al. Susceptibility to antifungal agents and enzymatic activity of Candida haemulonii and Cutaneotrichosporon dermatis isolated from soft corals on the Brazilian reefs. Arch Microbiol. 2016;198(10):963–971.
- Shu J, Ning P, Guo T, et al. First report of leaf spot caused by Colletotrichum fructicola and C. siamense on Zizyphus mauritiana in Guangxi, China. Plant Dis. 2020;104(12):3256–3256.
- Chai AL, Zhao Q, Li XJ, et al. First report of cercospora leaf spot caused by Cercospora cf. flagellaris on okra in China. Plant Dis. 2021;105(7):2018.
- Sarnecka AK, Nawrat D, Piwowar M, et al. DNA extraction from FFPE tissue samples – a comparison of three procedures. Contemp Oncol. 2019;23(1):52–58.
- Das P, Pandey P, Harishankar A, et al. A high yield DNA extraction method for medically important candida species: a comparison of manual versus QIAcube-based automated system. Indian J Med Microbiol. 2016;34(4):533–535.
- Malentacchi F, Ciniselli CM, Pazzagli M, et al. Influence of pre-analytical procedures on genomic DNA integrity in blood samples: the SPIDIA experience. Clin Chim Acta. 2015;440:205–210.
- Nasiri H, Forouzandeh M, Rasaee MJ, et al. Modified salting-out method: high-yield, high-quality genomic DNA extraction from whole blood using laundry detergent. J Clin Lab Anal. 2005;19(6):229–232.
- Lang J, Zhu R, Sun X, et al. Evaluation of the MGISEQ-2000 sequencing platform for Illumina target capture sequencing libraries. Front Genet. 2021;12:730519.
- Kong N, Ng W, Thao K, et al. Automation of PacBio SMRTbell NGS library preparation for bacterial genome sequencing. Stand Genomic Sci. 2017;12:27.
- Zhang L-L, Huang W, Zhang Y-Y, et al. Genomic and transcriptomic study for screening genes involved in the limonene biotransformation of Penicillium digitatum DSM 62840. Front Microbiol. 2020;11:744.
- Bickhart DM, McClure JC, Schnabel RD, et al. Symposium review: advances in sequencing technology herald a new frontier in cattle genomics and genome-enabled selection. J Dairy Sci. 2020;103(6):5278–5290.
- Liu L, Li Y, Li S, et al. Comparison of next-generation sequencing systems. J Biomed Biotechnol. 2012;2012:251364.
- Yoshinaga Y, Daum C, He G, et al. Genome sequencing. Methods Mol Biol. 2018;1775:37–52.
- Takashima M, Sriswasdi S, Manabe R-I, et al. A trichosporonales genome tree based on 27 haploid and three evolutionarily conserved 'natural' hybrid genomes. Yeast. 2018;35(1):99–111.
- Close D, Ojumu J. Draft genome sequence of the oleaginous yeast Cryptococcus curvatus ATCC 20509. Genome Announc. 2016;4(6):e01235–16.
- Sun S, Coelho MA, Heitman J, et al. Convergent evolution of linked mating-type loci in basidiomycete fungi. PLOS Genet. 2019;15(9):e1008365.
- Hofmeyer T, Hackenschmidt S, Nadler F, et al. Draft genome sequence of Cutaneotrichosporon curvatus DSM 101032 (formerly Cryptococcus curvatus), an oleaginous yeast producing polyunsaturated fatty acids. Genome Announc. 2016;4(3):e00362–16.
- Alva A, Sabido-Ramos A, Escalante A, et al. New insights into transport capability of sugars and its impact on growth from novel mutants of Escherichia coli. Appl Microbiol Biotechnol. 2020;104(4):1463–1479.
- Sharma NK, Behera S, Arora R, et al. Xylose transport in yeast for lignocellulosic ethanol production: current status. J Biosci Bioeng. 2018;125(3):259–267.
- Vasylyshyn R, Kurylenko O, Ruchala J, et al. Engineering of sugar transporters for improvement of xylose utilization during high-temperature alcoholic fermentation in Ogataea polymorpha yeast. Microb Cell Fact. 2020;19(1):96.
- Zhang B, Zhang J, Wang D, et al. Simultaneous fermentation of glucose and xylose at elevated temperatures co-produces ethanol and xylitol through overexpression of a xylose-specific transporter in engineered Kluyveromyces marxianus. Bioresour Technol. 2016;216:227–237.
- Runquist D, Fonseca C, Rådström P, et al. Expression of the Gxf1 transporter from Candida intermedia improves fermentation performance in recombinant xylose-utilizing Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2009;82(1):123–130.
- Young E, Poucher A, Comer A, et al. Functional survey for heterologous sugar transport proteins, using Saccharomyces cerevisiae as a host. Appl Environ Microbiol. 2011;77(10):3311–3319.
- Wang X, Goh E-B, Beller HR, et al. Engineering E. coli for simultaneous glucose-xylose utilization during methyl ketone production. Microb Cell Fact. 2018;17(1):12.
- Hua Y, Wang J, Zhu Y, et al. Release of glucose repression on xylose utilization in Kluyveromyces marxianus to enhance glucose-xylose co-utilization and xylitol production from corncob hydrolysate. Microb Cell Fact. 2019;18(1):24.
- Abe K, Uchida K. Correlation between depression of catabolite control of xylose metabolism and a defect in the phosphoenolpyruvate: mannose phosphotransferase system in Pediococcus halophilus. J Bacteriol. 1989;171(4):1793–1800.
- Khunnonkwao P, Jantama SS, Kanchanatawee S, et al. Re-engineering Escherichia coli KJ122 to enhance the utilization of xylose and xylose/glucose mixture for efficient succinate production in mineral salt medium. Appl Microbiol Biotechnol. 2018;102(1):127–141.
- Ranade S, Zhang Y, Kaplan M, et al. Metabolic engineering and comparative performance studies of Synechocystis sp. PCC 6803 strains for effective utilization of xylose. Front Microbiol. 2015;6:1484.
- Chen L, Zhang Z, Hoshino A, et al. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat Metab. 2019;1:404–415.
- Minard KI, McAlister-Henn L. Sources of NADPH in yeast vary with carbon source. J Biol Chem. 2005;280(48):39890–39896.
- Choi JW, Da Silva NA. Improving polyketide and fatty acid synthesis by engineering of the yeast acetyl-CoA carboxylase. J Biotechnol. 2014;187:56–59.
- Montoya AM, Luna-Rodríguez CE, Bonifaz A, et al. Physiological characterization and molecular identification of some rare yeast species causing onychomycosis. J Mycol Med. 2021;31(2):101121.
- Gallagher MD, Chen-Plotkin AS. The Post-GWAS era: from association to function. Am J Hum Genet. 2018;102(5):717–730.
- Price MN, Arkin AP. Curated BLAST for genomes. mSystems. 2019;4(2):e00072-19.
- Pace J, Youens-Clark K, Freeman C, et al. PuMA: a papillomavirus genome annotation tool. Virus Evol. 2020;6(2):veaa068.
- Magrini V, Gao X, Rosa BA, et al. Improving eukaryotic genome annotation using single molecule mRNA sequencing. BMC Genomics. 2018;19(1):172.
- Watson JD, Laskowski RA, Thornton JM, et al. Predicting protein function from sequence and structural data. Curr Opin Struct Biol. 2005;15(3):275–284.
- Skov LK, Mirza O, Henriksen A, et al. Amylosucrase, a glucan-synthesizing enzyme from the alpha-amylase family. J Biol Chem. 2001;276(27):25273–25278.
- Dietmann S,PJ, Notredame C, Heger A, et al. A fully automatic evolutionary classification of protein folds: dali domain dictionary version 3. Nucleic Acids Res. 2001;29(1):55–57.
- Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60(12):2256–2268.
- Harrison A, Pearl F, Sillitoe I, et al. Recognizing the fold of a protein structure. Bioinformatics. 2003;19(14):1748–1759.
- Binkowski TA, Freeman P, Liang J, et al. pvSOAR: detecting similar surface patterns of pocket and void surfaces of amino acid residues on proteins. Nucleic Acids Res. 2004;32:W555–8.
- Ra L. SURFNET: a program for visualizing molecular surfaces, cavities, and intermolecular interactions. J Mol Graph. 1995;13(5):323–330.
- Glaser F, Morris RJ, Najmanovich RJ, et al. A method for localizing ligand binding pockets in protein structures. Proteins. 2006;62(2):479–488.
- Innis CA, Anand AP, Sowdhamini R, et al. Prediction of functional sites in proteins using conserved functional group analysis. J Mol Biol. 2004;337(4):1053–1068.
- Wilkins A, Erdin S, Lua R, et al. Evolutionary trace for prediction and redesign of protein functional sites. Methods Mol Biol. 2012;819:29–42.
- Pillai-Kastoori L, Schutz-Geschwender AR, Harford JA, et al. A systematic approach to quantitative western blot analysis. Anal Biochem. 2020;593:113608.
- Atout S, Shurrab S, Loveridge C, et al. Evaluation of the suitability of RNAscope as a technique to measure gene expression in clinical diagnostics: a systematic review. Mol Diagn Ther. 2022;26(1):19–37.,
- Aslam B, Basit M, Nisar MA, et al. Proteomics: technologies and their applications. J Chromatogr Sci. 2017;55(2):182–196.
- Pappireddi N, Martin L, Wühr M, et al. A review on quantitative multiplexed proteomics. Chembiochem. 2019;20(10):1210–1224.