
Abstract
Aspergillus sojae has long been considered a domesticated strain of Aspergillus parasiticus. This study delineated relationships among the two species and an Aspergillus PWE36 isolate. Of 25 examined clustered aflatoxin genes of PWE36, 20 gene sequences were identical to those of A. sojae, but all had variations to those of A. parasiticus. Additionally, PWE36 developmental genes of conidiation and sclerotial formation, overall, shared higher degrees of nucleotide sequence identity with A. sojae genes than with A. parasiticus genes. Examination of defective cyclopiazonic acid gene clusters revealed that the PWE36 deletion pattern was identical only to those of A. sojae. Using A. sojae SMF134 genome sequence as a reference, visualization of locally collinear blocks indicated that PWE36 shared higher genome sequence homologies with A. sojae than with A. parasiticus. Phylogenetic inference based on genome-wide single nucleotide polymorphisms (SNPs) and total SNP counts showed that A. sojae strains formed a monophyletic clade and were clonal. Two (Argentinian and Ugandan) A. parasiticus isolates but not including an Ethiopian isolate formed a monophyletic clade, which showed that A. parasiticus population is genetically diverse and distant to A. sojae. PWE36 and A. sojae shared a most recent common ancestor (MRCA). The estimated divergence time for PWE36 and A. sojae was about 0.4 mya. Unlike Aspergillus oryzae, another koji mold that includes genetically diverse populations, the findings that current A. sojae strains formed a monophyletic group and shared the MRCA with PWE36 allow A. sojae to be continuously treated as a species for food safety reasons.
1. Introduction
Aspergillus sojae, like Aspergillus oryzae, is a highly valued koji (starter) mold used to produce fermented foods, such as miso and soy sauce [Citation1]. It together with Aspergillus parasiticus, A. oryzae, and Aspergillus flavus is part of the genus’ section Flavi (“section” is a taxonomic rank in-between genus and species) [Citation2]. Non-aflatoxigenic A. sojae is commonly believed to be a domesticated A. parasiticus strain. This is because, unlike A. oryzae, it has only been isolated from koji [Citation3]. A. parasiticus, which produces aflatoxins B1, B2, G1 and G2, and A. sojae are morphologically similar although they can be differentiated by conidial diameter and colony color [Citation4]. These phenotypic differences, however, are subtle and intergradation often exists between them. Kurtzman et al. reported that DNA homology between A. sojae and A. parasiticus was 91%, and it was 100% between A. oryzae and A. flavus. Hence, they proposed that A. sojae, A. parasiticus, and A. oryzae should be reduced to varietal status [Citation5]. One school of thought also holds that traits gained during domestication caused the koji molds to lose morphological and physiological characteristics that at present still persist in wild-type A. parasiticus and A. flavus.
Several atypical A. parasiticus isolates that produced B- and G-type aflatoxins were collected from pistachio nut fruits and designated as PWE (for Pistachio Winter Experiment) strains [Citation6]. However, distinct colony morphological variations exist among them. Their morphologies also exhibit similarity and dissimilarity to those of A. parasiticus and A. sojae reference strains. The PWE isolates at the molecular level have a unique HA-coding insertion (CTCATG) in the aflatoxin pathway regulatory gene, aflR, which is identical to what has been known for A. sojae isolates [Citation7]. This insertion is not present in A. parasiticus aflR. How close the PWE isolates are to A. sojae and to A. parasiticus was unclear. The presence or absence of the CTCATG sequence and nucleotides at position #1154 in aflR are sufficient for Aspergillus species identification. For example, these molecular features correctly reclassified non-aflatoxigenic A. oryzae ATCC18895 to A. sojae [Citation8].
In the genomics era, high-throughput whole genome sequencing has become a routine. Genome sequence information for a fungal isolate now can be readily obtained in days. This has made molecular classification techniques employed in the past; for example, DNA complementarity, electrophoretic karyotypes, restriction fragment length polymorphism (RFLP), and random amplified polymorphic DNA (RAPD) become obsolete. Accurate identification of fungal species no longer relies on nucleotide sequences of polymerase chain reaction (PCR) fragments of coding or rRNA gene(s) [Citation9]. Additionally, culture characteristics ancillary to the sequence techniques are affected by environmental and nutritional conditions, which makes it difficult to score morphological data without error. Misclassification of fungal isolates therefore is not uncommon [Citation8,Citation10].
In the present study, we sequenced the genome of a representative PWE36 strain. We first compared PWE36 genes, which included 25 clustered genes involved in aflatoxin biosynthesis and 23 key developmental genes associated with conidiation (13 genes) and sclerotial formation (10 genes), to the orthologous genes of A. sojae and A. parasiticus. We also explored their genetic relatedness based on deletion patterns in the cyclopiazonic acid (CPA) gene clusters. We further provided visualization of whole genome homologies using the well-assembled A. sojae SMF134 genome [Citation11] as the reference. Lastly, we used genome-wide single nucleotide polymorphisms (SNPs) to assess the evolutionary origin of A. sojae and to delineate phylogenetic relationships among A. sojae, PWE36, and A. parasiticus.
2. Materials and methods
2.1. Fungal strains and assembled genome sequences
lists A. sojae strains and A. parasiticus isolates, geographic locations of collection, and assembled genome sequences used in this study. The genome sequences were retrieved from the National Center for Biotechnology Information (NCBI) genome database (https://www.ncbi.nlm.nih.gov/genome/) with respective species names.
Table 1. Aspergillus genome sequences used in this study.
2.2. Genome sequencing and assembly of Aspergillus PWE36
Aspergillus PWE36 is one of the several aflatoxigenic isolates collected at Wolfskill Grant Experimental Farm (University of California Davis, Winters, California, USA) [Citation6]. Their colony morphology and conidial surface texture resemble to those of A. sojae and A. parasiticus. PWE36 was selected as the representative strain for genome sequencing. It was grown in yeast peptone dextrose (YPD) broth at 28 °C for 24 h. Harvested mycelia were frozen and grounded to a fine powder with a pestle and mortar. Fungal DNA was extracted using a MasterPure DNA Purification Kit (Epicentre® Biotechnologies, Madison, Wisconsin, USA). Genome sequencing was completed through a service agreement with CosmosID (Germantown, Maryland, USA). The libraries were sequenced using a whole-genome shotgun approach on an Illumina MiSeq instrument. The genome coverage was 40×. Sequence reads were first quality controlled using BBDuk. Trimmed FASTA files were then subjected to reference guided metagenomic assembly using SPAdes [Citation12]. A total of 1,434 contigs (N50 is 53,221) were assembled. The BioProject is PRJNA575261, and the genome sequence is under Accession number WELH00000000.1.
2.3. Sequence alignments of clustered aflatoxin genes and developmental genes
Twenty-five genes in the aflatoxin gene cluster of A. parasiticus SU-1 [Citation13] served as the original sequence search templates (). Its GenBank accession number is AY371490. BlastN search against PWE36 genome assembly, GCA_019419805.1, was carried out first and corresponding gene sequences were obtained. The PWE36 gene sequences were then used in BlastN search against the assembled genome sequences of A. parasiticus and A. sojae (). Similarly, selected developmental genes related to conidiation and sclerotial production were first identified from A. flavus NRRL3357 () and served as BlastN search query sequences against PWE36 genome assembly. The obtained PWE36 gene sequences were then used in BlastN search against the assembled genome sequences of A. parasiticus and A. sojae.
Table 2. Degrees of sequence identity of PWE36 aflatoxin biosynthesis genes to those of Aspergillus sojae strains and Aspergillus parasiticus isolates.
Table 3. Degrees of sequence identity of PWE36 developmental genes to those of Aspergillus sojae strains and Aspergillus parasiticus isolates.
2.4. Characterization of pksA genes and aflR genes of A. parasiticus, PWE36, and A. sojae
The polyketide synthase gene, pksA/aflC, and the aflatoxin pathway-specific regulatory gene, aflR, of A. sojae SRRC1123 (GenBank accession numbers: AY607769 and KT829484) were retrieved from the NCBI nucleotide database (https://www.ncbi.nlm.nih.gov/nuccore/?term=). The sequence portions containing the desired nucleotide substitutions and insertion/deletion were identified and used to align the assembled genome sequences of A. parasiticus and A. sojae () with BlastN search.
2.5. Characterization of defective CPA gene clusters of A. parasiticus, PWE36, and A. sojae
Unlike A. flavus that produces CPA, A. sojae and A. parasiticus are unable to produce this metabolite because their CPA gene clusters are defective [Citation14,Citation15]. For determining deletion patterns of the CPA gene clusters in A. sojae and A. parasiticus, the 16.8 kb CPA gene cluster of A. flavus AF36 (GenBank accession number: JN712209) was used as the alignment template in BlastN search against genome sequences (). Localization and detailed analysis of deleted sequence regions were performed manually.
2.6. Visualization of genome sequence homology and phylogeny of A. sojae, PWE36, and A. parasiticus
Mauve, an online system for aligning multiple genome sequences was downloaded (http://darlinglab.org/mauve/mauve.html) [Citation16]. Its program “progressiveMauve” was used to extract nucleotide variations that exist between compared genomes. For visual representation of two genome sequences aligned, the program “Move Contigs” of Mauve was used to draw locally collinear blocks (LCBs), which show homologous regions between compared genome assemblies. A. sojae SMF134 genome sequence, which has been assembled into eight chromosomes [Citation11], was used as the reference. Phylogenetic inference with concatenated total SNP sequences was performed by the weighted Neighbor-Joining method of the online program MAFFT (version 7) (https://mafft.cbrc.jp/alignment/server/phylogeny.html) [Citation17]. Aligning genome sequences, extracting SNPs of aligned paired or all genomes, filtering out noise (i.e., gaps and ambiguous bases), concatenating cleaned SNPs, and converting sequences to FASTA format were carried out using a custom JavaScript [Citation18]. A phylogeny with rectangular tree layout was prepared using FigTree v1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/).
3. Results and discussion
3.1. Comparison of aflatoxin biosynthesis genes and those associated with development
In contrast to A. parasiticus, which is aflatoxigenic, A. sojae does not produce aflatoxins. Although aflatoxins (B1, B2, G1 and G2) are not essential for growth and development of producing fungi, the gene cluster is believed to have been maintained for 25 million years [Citation19], which suggests that producing these polyketide-derived metabolites in nature has an adaptive value. In this study, the comparison of 25 genes in the aflatoxin gene clusters showed that nucleotide sequences, which include exons and introns, of 20 genes between PWE36 and the five A. sojae strains were identical (). In contrast, all 25 genes between PWE36 and the A. parasiticus isolates collected from different geographical locations had minor sequence variations. Consistently, developmental genes of PWE36 associated with conidiation shared higher degrees of nucleotide sequence identity with A. sojae genes than with A. parasiticus genes. But three of the 10 PWE36 developmental genes associated with sclerotial formation, veA [Citation20,Citation21], laeA [Citation22] and sfgA [Citation23], had higher sequence variations when compared to A. sojae genes than to A. parasiticus genes. Thousands of years’ use of koji for preparation of East Asian traditional fermented foods has resulted in morphological changes of A. oryzae and A. sojae. For example, A. oryzae has sparse sporulation, floccose aerial mycelia and few or no sclerotia, and aging colonies change color toward brown rather than green as seen for A. flavus and A. parasiticus [Citation24,Citation25]. Floccose mycelia and aged color change also are reported for A. sojae. It is not known whether the selection pressure from the solid-state cultivation during fermentation processes, which enables fast growth and conidiation, forces the domesticated koji molds to adapt to specific production environments and affects formation of sclerotia, another type of propagules produced under unfavorable growth conditions.
3.2. SNPs in pksA genes and indels in aflR genes of A. parasiticus, PWE36, and A. sojae
Functional pksA/aflC and aflR genes are required for aflatoxin production in A. parasiticus [Citation26,Citation27]. Sequence comparison has indicated that both orthologous genes in A. sojae are defective [Citation7,Citation28], thereby rendering A. sojae unable to produce aflatoxins. In this study, the nucleotides of pksA/aflC genes at positions #6127 and #6133 for all A. parasiticus isolates and all A. sojae strains examined were “GC” and “AA”, respectively, which are consistent with previous findings (). Notably, PWE36 had intermediate nucleotide changes. At position #6127 it had “A” identical to that in A. sojae, but at position #6133 it had “C” identical to that in A. parasiticus. The acquisition of “A” in position #6133 in A. sojae, which results in a pre-termination codon, likely occurred after PWE36-like ancestor (and A. sojae) diverged from A. parasiticus. Probably the mutation was introduced during domestication. Similarly, the insertion (duplication) of the CTCATG nucleotides in aflR of PWE36 indicated its divergence from A. parasiticus (). The ensuring change of nucleotide “C” at position #1154 to “T” in A. sojae further suggests a genetic transition from a PWE36-like ancestor to A. sojae.
Figure 1. Alignment of portions of pksA/aflC sequences (A); aflR sequences (B) from Aspergillus parasiticus isolates, PWE36, and Aspergillus sojae strains. SNPs in blue that yield pre-termination codons in pksA/aflC (TAA) and aflR (TGA) genes, respectively, are yellow highlighted. Numbers correspond to nucleotide positions in A. sojae gene sequences.

Historically speaking, use of koji in food fermentation began first in China over 2,000 to 3000 years ago, and the technology was adopted by other regions such as Japan and Korea during different periods [Citation25,Citation29]. To investigate whether PWE36-like Aspergillus exists in regions(s) other than California, we therefore searched the NCBI databases for possible candidates. To our surprise, A. parasiticus OPS651, isolated from peanut in China and producing B- and G-type aflatoxins [Citation30], also contained the additional HA-coding sequence, CTCATG. It is probable that POS651 is genetically close to PWE36. As more field Aspergillus isolates having molecular features identical to PWE36 are collected and their genomes sequenced, we would have a better understanding of the phylogenetic relationship of A. parasiticus and A. sojae.
3.3. Defective CPA gene clusters of A. parasiticus, PWE36, and A. sojae
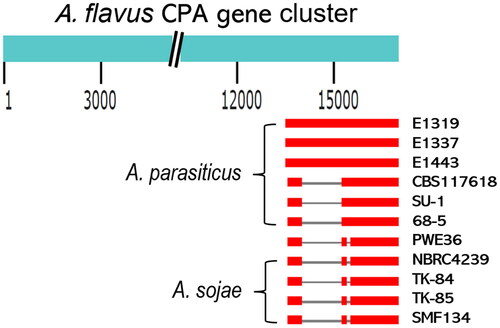
A. parasiticus and A. sojae, unlike A. flavus and A. oryzae, are unable to produce CPA because they do not have a complete CPA gene cluster, which includes at least three characterized genes, maoA, dmaT and pks-nrps/cpaA [Citation3,Citation14,Citation15]. The exact deletion(s) in A. parasiticus and A. sojae CPA gene clusters had not yet been determined. To examine the relationship of PWE36 with A. parasiticus and with A. sojae, we defined deletion patterns in their CPA gene clusters. Using the complete A. flavus CPA gene cluster as a reference, we found that only about 3.5 kb in the 3′ portion that harbors the pks-nrps/cpaA gene was retained in the incomplete CPA gene clusters of the Ethiopian A. parasiticus isolates (). Comparatively, A. parasiticus isolates CBS117618, SU-1, and 68-5 [Citation31] had a further 1.2 kb deletion. On closer manual examination, we found that the corresponding 1.2 kb region in these isolates was replaced by a 7.7 kb nonhomologous region (data not shown). The CPA gene clusters of the four A. sojae strains and PWE36 had the same 7.7 kb replacement; all also had an additional short 112 bp deletion. The transition of these specific deletion patterns suggests that PWE36 is genetically closer to A. sojae than to A. parasiticus.
Figure 2. Schematic representations of deletions in the CPA gene clusters of Aspergillus parasiticus isolates, PWE36, and Aspergillus sojae strains. The CPA gene cluster of Aspergillus flavus AF36 (16.8 kb) was used as the alignment template. The comparisons on the lower panel were drawn to scale. Blank space indicates deleted portions. Grey lines are nonhomologous regions of replacement and the 112 bp deletions. Thick red lines are highly homologous sequences.
3.4. Visualization of genome-wide sequence homology
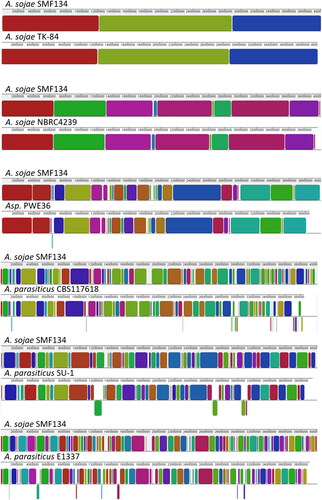
To visualize sequence homologies among the genomes of A. sojae, PWE36, and A. parasiticus, we used the A. sojae SMF134 genome sequence as a reference, whose eight chromosomes had been assembled [Citation11]. The alignment template was based on concatenated sequences of the chromosomes. In , homologous genome sequence regions were indicated by colored collinear blocks (LCBs); the larger the LCBs present the wider the homologous regions are. As revealed, genome sequences of A. sojae SMF134, TK-84, and NBRC4329 were highly homologous. In contrast, the SMF134 genome sequence shared much smaller LCBs with those of the three A. parasiticus isolates. Additionally, we found that various sequence translocations were present in the A. parasiticus genomes. Overall, the PWE36 genome sequence shared higher degrees of homology with those of A. sojae strains than with the A. parasiticus isolates.
Figure 3. Schematic representation of homologous regions in genome sequences of Aspergillus sojae strains, PWE36, and Aspergillus parasiticus isolates. The genome sequence of A. sojae SMF134 that had been assembled at the chromosome level was used as the reference; it was concatenated in chromosome order (from I to VIII). Colored blocks are locally collinear blocks (LCBs), which indicate homologous regions between two genomes. The LCB weight was set at around 550 except for the SMF134/TK-84 pair, of which the lowest score was 24,283. Regions that were split and translocated in a compared genome are shown as inverted segments at the bottom.
3.5. SNP counts of paired genome sequence alignments and total SNP-based phylogeny
In comparison to A. oryzae, of which over 200 genome sequences are publicly available, only a few A. sojae and less than 10 A. parasiticus genome sequences have been deposited to NCBI. Of the three A. sojae strains, SMF134 and TK-84 [Citation11,Citation29] shared the lowest total SNP count (2840) with each other (). NBRC4239 [Citation15] also shared low total SNP counts with SM134 and TK-84, respectively. These low SNP counts suggest that current A. sojae strains are clonal. Low total SNP counts of A. oryzae clonal strains such as 3.042 and 100-8 (=1246) from China [Citation32,Citation33] as well as SRCM101975 and SRCM101989 from South Korea (=379) [Citation34] also have been reported. Total SNP counts between the Ethiopian A. parasiticus isolate (E1337) and either CBS117618 or SU-1 were high although the count shared by CBS117618 and SU-1, collected respectively from the leaf of an Argentinian wild peanut species and a Ugandan peanut [Citation35,Citation36], was only about half. It is not clear whether geographic separation and niche adaptation have a bearing on the markedly different evolutionary distance. Alternately, E1337 might be a species very closely related to A. parasiticus. The total SNP counts among A. parasiticus isolates varied greatly () and can be translated into approximately 98.6% to 99.3% genome sequence identity. Isolates of A. flavus (S- and L-morphotypes) and A. oryzae share overall total SNP counts of 220,000 and 300,000, respectively [Citation34]. Hence, A. parasiticus has higher genetic diversity than A. flavus and A. oryzae (∼99.2% to 99.4% genome sequence identity) despite that a large group of A. oryzae has been shown to be phylogenetically closer to A. aflatoxiformans than to A. flavus [Citation2,Citation18,Citation37]. PWE36 total SNP counts shared with the three A. sojae strains were in that range reported for A. flavus and A. oryzae. They were about four- to 10-fold of those shared among the A. sojae strains. The total SNP counts shared by PWE36 and the A. sojae strains were only half of those shared by PWE36 and the A. parasiticus isolates (), which indicates close genetic relatedness of the former aspergilli.
Figure 4. (A) Total SNP counts from paired genome sequence comparisons among PWE36, Aspergillus sojae strains, and Aspergillus parasiticus isolates; (B) Phylogenetic tree inferred from concatenated sequences of total SNPs by the Neighbor-Joining method with 100 bootstrap iterations. The number of total SNP count for each sequence was 618,048. The estimated divergence time (1.1 mya) between A. parasiticus SU-1 and A. sojae NRBC4239 was derived from the reference divergence time scale (3.8 mya) between Aspergillus flavus NRRL3357 and Aspergillus oryzae RIB40 [Citation38].
![Figure 4. (A) Total SNP counts from paired genome sequence comparisons among PWE36, Aspergillus sojae strains, and Aspergillus parasiticus isolates; (B) Phylogenetic tree inferred from concatenated sequences of total SNPs by the Neighbor-Joining method with 100 bootstrap iterations. The number of total SNP count for each sequence was 618,048. The estimated divergence time (1.1 mya) between A. parasiticus SU-1 and A. sojae NRBC4239 was derived from the reference divergence time scale (3.8 mya) between Aspergillus flavus NRRL3357 and Aspergillus oryzae RIB40 [Citation38].](/cms/asset/9611a16f-81bc-4e09-a194-705029b4f653/tmyb_a_2217495_f0004_c.jpg)
3.6. Divergence of current A. sojae strains from PWE36
A phylogenetic tree inferred by the Neighbor-Joining (NJ) method, using the concatenated sequences of total SNPs, was consistent with those inferred based on total SNP counts. For example, A. sojae SMF134, NBRC4239, and TK-84 comprised a monophyletic clade. A. parasiticus CBS117618 and SU-1 grouped in a separate monophyletic clade (). Notably, PWE36 shared a most recent common ancestor (MRCA) with the A. sojae strains and were phylogenetically distant from the three A. parasiticus isolates. A study inferring evolutionary relationships of Aspergillus species based on 1668 Benchmarking Universal Single-Copy Ortholog (BUSCO) genes has concluded that A. parasiticus (SU-1) and A. sojae (NRBC4239) diverged about 1.1 million years ago [Citation38]. We used the phylogenetic tree derived from total SNP counts and extrapolated that current A. sojae strains diverged from Aspergillus PWE36 roughly 430 thousand years ago. PWE36 was much more distant from Ethiopian A. parasiticus isolates (about 1.4 million years ago) than from Ugandan and Argentinian A. parasiticus isolates. Nonetheless, PWE36 divergence from A. parasiticus occurred much more recent than the 3.8 million years ago estimated for A. flavus (NRRL3357) and A. oryzae (RIB40) [Citation38].
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Ito K, Matsuyama A. Koji molds for Japanese soy sauce brewing: characteristics and key enzymes. J Fungi. 2021;7(8):658.
- Frisvad JC, Hubka V, Ezekiel CN, et al. Taxonomy of Aspergillus section flavi and their production of aflatoxins, ochratoxins and other mycotoxins. Stud Mycol. 2019;93:1–63.
- Kim KM, Lim J, Lee JJ, et al. Characterization of Aspergillus sojae isolated from Meju, Korean traditional fermented soybean brick. J Microbiol Biotechnol. 2017;27(2):251–261.
- Klich MA. Identification of common Aspergillus species. Utrecht: Centraalbureau voor Schimmelcultures, 2002.
- Kurtzman CP, Smiley MJ, Robnett CJ, et al. DNA relatedness among wild and domesticated species in the Aspergillus flavus group. Mycologia. 1986;78(6):955–959.
- Hua SST, Parfitt DE, Sarreal SBL, et al. First report of an atypical new Aspergillus parasiticus isolates with nucleotide insertion in aflR gene resembling to A. sojae. Mycotoxin Res. 2018;34(2):151–157.
- Takahashi T, Chang P-K, Matsushima K, et al. Nonfunctionality of Aspergillus sojae aflR in a strain of Aspergillus parasiticus with a disrupted aflR gene. Appl Environ Microbiol. 2002;68(8):3737–3743.
- Watson AJ, Fuller LJ, Jeenes DJ, et al. Homologs of aflatoxin biosynthesis genes and sequence of aflR in Aspergillus oryzae and Aspergillus sojae. Appl Environ Microbiol. 1999;65(1):307–310.
- Arias RS, Orner VA, Martinez-Castillo J, et al. Aspergillus section flavi, need for a robust taxonomy. Microbiol Resour Announc. 2021;10(48):e0078421.
- Houbraken J, Visagie CM, Frisvad JC. Recommendations to prevent taxonomic misidentification of genome-sequenced fungal strains. Microbiol Resour Announc. 2021;10(48):e0107420.
- Kim KU, Kim KM, Choi YH, et al. Whole genome analysis of Aspergillus sojae SMF 134 supports its merits as a starter for soybean fermentation. J Microbiol. 2019;57(10):874–883.
- Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–477.
- Yu J, Chang P-K, Ehrlich KC, et al. Clustered pathway genes in aflatoxin biosynthesis. Appl Environ Microbiol. 2004;70(3):1253–1262.
- Chang P-K, Horn BW, Dorner JW. Clustered genes involved in cyclopiazonic acid production are next to the aflatoxin biosynthesis gene cluster in Aspergillus flavus. Fungal Genet Biol. 2009;46(2):176–182.
- Sato A, Oshima K, Noguchi H, et al. Draft genome sequencing and comparative analysis of Aspergillus sojae NBRC4239. DNA Res. 2011;18(3):165–176.
- Darling AC, Mau B, Blattner FR, et al. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–1403.
- Katoh K, Misawa K, Kuma K, et al. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059–3066.
- Chang P-K, Chang TD, Katoh K. Deciphering the origin of Aspergillus flavus NRRL21882, the active biocontrol agent of Afla-Guard. Lett Appl Microbiol. 2021;72(5):509–516.
- Ehrlich KC, Yu J, Cotty PJ. Aflatoxin biosynthesis gene clusters and flanking regions. J Appl Microbiol. 2005;99(3):518–527.
- Calvo AM, Bok J, Brooks W, et al. veA is required for toxin and sclerotial production in Aspergillus parasiticus. Appl Environ Microbiol. 2004;70(8):4733–4739.
- Chang P-K, Scharfenstein LL, Li P, et al. Aspergillus flavus VelB acts distinctly from VeA in conidiation and may coordinate with FluG to modulate sclerotial production. Fungal Genet Biol. 2013;58–59:71–79.
- Kale SP, Milde L, Trapp MK, et al. Requirement of LaeA for secondary metabolism and sclerotial production in Aspergillus flavus. Fungal Genet Biol. 2008;45(10):1422–1429.
- Yuan XY, Li JY, Zhi QQ, et al. SfgA renders Aspergillus flavus more stable to the external environment. J Fungi. 2022;8(6):638.
- Jorgensen TR. Identification and toxigenic potential of the industrially important fungi, Aspergillus oryzae and Aspergillus sojae. J Food Prot. 2007;70(12):2916–2972.
- Machida M, Yamada O, Gomi K. Genomics of Aspergillus oryzae: learning from the history of koji mold and exploration of its future. DNA Res. 2008;15(4):173–183.
- Feng GH, Leonard TJ. Characterization of the polyketide synthase gene (pksL1) required for aflatoxin biosynthesis in Aspergillus parasiticus. J Bacteriol. 1995;177(21):6246–6254.
- Chang P-K, Ehrlich KC, Yu J, et al. Increased expression of Aspergillus parasiticus aflR, encoding a sequence-specific DNA-binding protein, relieves nitrate inhibition of aflatoxin biosynthesis. Appl Environ Microbiol. 1995;61(6):2372–2377.
- Chang P-K, Matsushima K, Takahashi T, et al. Understanding nonaflatoxigenicity of Aspergillus sojae: a windfall of aflatoxin biosynthesis research. Appl Microbiol Biotechnol. 2007;76(5):977–984.
- Watarai N, Yamamoto N, Sawada K, et al. Evolution of Aspergillus oryzae before and after domestication inferred by large-scale comparative genomic analysis. DNA Res. 2019;26(6):465–472.
- Garber NP, Cotty PJ. Aspergillus parasiticus communities associated with sugarcane in the Rio Grande Valley of Texas: implications of global transport and host association within Aspergillus section flavi. Phytopathology. 2014;104(5):462–471.
- Faustinelli PC, Wang XM, Palencia ER, et al. Genome sequences of eight Aspergillus flavus spp. and one A. parasiticus sp., isolated from peanut seeds in Georgia. Genome Announc. 2016;4(2):e00278–e00216.
- Zhao G, Yao Y, Hou L, et al. Draft genome sequence of Aspergillus oryzae 100-8, an increased acid protease production strain. Genome Announc. 2014;2(3):e00548–14.
- Zhao G, Yao Y, Qi W, et al. Draft genome sequence of Aspergillus oryzae strain 3.042. Eukaryot Cell. 2012;11(9):1178.
- Chang P-K. Genome-wide nucleotide variation distinguishes Aspergillus flavus from Aspergillus oryzae and helps to reveal origins of atoxigenic A. flavus biocontrol strains. J Appl Microbiol. 2019;127(5):1511–1520.
- Pildain MB, Frisvad JC, Vaamonde G, et al. Two novel aflatoxin-producing Aspergillus species from Argentinean peanuts. Int J Syst Evol Microbiol. 2008;58(3):725–735.
- Linz JE, Wee J, Roze LV. Aspergillus parasiticus SU-1 genome sequence, predicted chromosome structure, and comparative gene expression under aflatoxin-inducing conditions: evidence that differential expression contributes to species phenotype. Eukaryot Cell. 2014;13(8):1113–1123.
- Kjærbølling I, Vesth T, Frisvad JC, et al. A comparative genomics study of 23 Aspergillus species from section flavi. Nat Commun. 2020;11(1):1106.
- Steenwyk JL, Shen XX, Lind AL, et al. A robust phylogenomic time tree for biotechnologically and medically important fungi in the genera Aspergillus and Penicillium. mBio. 2019;10(4):e00925–19.