
Abstract
The symbiotic association between fungus-gardening termites Macrotermes and its fungal symbiont has a moderate degree of specificity—although the symbiotic fungi (Termitomyces) form a monophyletic clade, there is not a one-to-one association between termite species and their fungus-garden associates. Here, we aim to determine the origin and phylogenetic relationships of Termitomyces in Oman. We used sequences of the internal transcribed spacer region (ITS) and the nuclear large subunit ribosomal RNA (LSU rRNA, 25S) gene and analyzed these with sequences of Termitomyces from other geographic areas. We find no evidence for more than a single colonization of Oman by Termitomyces. Unexpectedly, we find Termitomyces in Oman is most closely related to the symbiont of M. subhyalinus in West Africa rather than to those of geographically closer populations in East Africa.
Keywords:
1. Introduction
Fungus-gardening termites (Termitidae: Macrotermitinae) have obligate symbioses with the fungus Termitomyces (Basidiomycete: Agaricales: Lyophyllaceae). Fungus gardens inside the termite nest are provided with buffered temperature and humidity conditions, and the fungus provides food for its host termites. Termitophilic fungi are cultivated on plant-based substrates, called “fungus combs” (), made of termite fecal matter from pre-digested plant parts (e.g. leaf litter, wood and grass) contained in special chambers inside the termite nest [Citation1,Citation2]. The fungus produces nodules, vegetative spherical growths about 0.1–2 mm in diameter (), on the comb surface that serve as a food source for termite workers [Citation3].
Figure 1. Fungus comb in fungus garden of Macrotemes subhyalinus in Oman, yellow arrow showing fungus nodules. Photo by Hilal Al-Shamakhi.

Unlike the fungi cultivated by attine ants, in which some species of fungus have close free-living close relatives [Citation4–7], no Termitomyces are known to have a free-living state and they form a monophyletic group in phylogenetic analyses [Citation8]. Fungus-gardening termites (Macrotermitinae) also form a monophyletic group [Citation9–11] and all species cultivate Termitomyces as a food source [Citation12]. Together, these observations imply a single origin of the termite-fungus mutualism: the relationship between termites and their cultivated fungus is symmetrical in that both partners have a single origin with no reversal to a non-symbiotic state, and both partners are obligatorily dependent on this relationship [Citation1,Citation8].
The evolutionary relationships between fungus-gardening termites and their mutualistic fungi have yet to be fully resolved, partly due to their high complexity. For example, even in cases of vertical transmission where fungi are thought to be inherited from parental colonies, the symbiotic fungi used by a particular termite lineage do not necessarily form a monophyletic group [Citation8,Citation13,Citation14]. For example, Macrotemes bellicosus has associations with four different lineages of Termitomyces that do not form a clade [Citation8,Citation15]. The pattern is consistent with horizontal transfer of the fungus between species [Citation15]. In contrast, high specificity is found in some other lineages despite the potential for horizontal transfer when spores are collected from the surrounding environment for inoculating the comb. For example, M. natalensis is associated with a single symbiont lineage of Termitomyces [Citation16,Citation17]. Overall, fungus-gardening termites show a range of specificity with respect to their Termitomyces symbionts, and apparent transmission mode does not unambiguously predict host-symbiont specificity.
To date, the Termitomyces association with M. subhyalinus in Oman have not been included in studies of the fungus-termite associations. Macrotermes subhyalinus occurs in the Dhofar region of southern Oman, a local center of endemism [Citation18], where it forms mounds of up to 2 m in diameter. Here, we sample populations of Termitomyces from across the distribution of M. subhyalinus in Oman to assess its diversity and to determine its phylogenetic relationship with Termitomyces from other regions.
2. Materials and methods
2.1. Specimen sampling
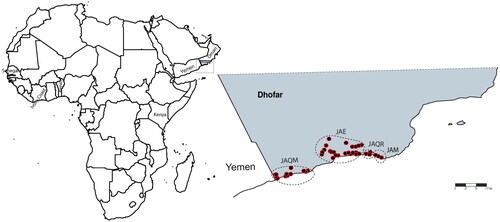
Fungal combs were collected from 76 colonies of M. subhyalinus from across Dhofar, southern Oman (; Supplementary Material Table S1) during June and July 2015. Nodules were separated from fungal combs and surface washed in 70% ethanol, then preserved in 99% ethanol until extraction of genomic DNA.
Figure 2. Map showing sampled localities across Dhofar region southern of Oman. (JAQM) Jabal qamar, (JAE) Jabal eitin, (JAQR) Jabal qara, and (JAM) Jabal murbat.
2.2. DNA extraction
A single nodule (2–3mm) was selected from each of several combs in each colony. Genomic DNA was extracted by grinding the samples from one nest manually using a pestle in 600 µl 2 M CTAB buffer and 10 µl Proteinase K. The mixture was then incubated on a hot plate for two hours at 55 °C before heating at 95 °C for 10 min. After cooling to room temperature, 300 µl of chloroform was added and the mixture was gently rocked for 15 min, followed by micro-centrifuging at 15800 RCF for 5 min. The supernatant was removed to a new tube and 600 µl of 100% isopropanol was added, vortexed and centrifuged at 15800 RCF for 10 min. After discarding the supernatant, the DNA pellet was washed with 500 µl of 80% ethanol and centrifuged at 15800 RCF for 1 min. The supernatant was removed and the pellet was air dried before being re-suspended in 50 µl sterile distilled water. To assess the integrity of the genomic DNA, 2 µl of template DNA was run through a 1% agarose gel at constant voltage V400/A120 for 60 min and visualized under ultraviolet light.
2.3. Amplification and sequencing protocol
We amplified the internal transcribed spacer region (ITS1-5.8S-ITS2) of the nuclear ribosomal RNA repeat regions and the D2-D3 expansion regions of the nuclear large subunit ribosomal RNA gene (LSU rRNA, 25S). About 670 bp of ITS was amplified with primers ITS1 (5′- TCC GTA GGT GAA CCT GCG G-3′) and ITS4 (5′-TCC TCC GCT TAT TGA TAT GC-3′) [Citation19] and PCR consisted of an initial denaturing step of 4 min at 94 °C, then 35 cycles of denaturing at 94 °C (30 s), annealing at 55 °C (30 s) and extension at 72 °C (90 s), followed by an additional extension at 72 °C (10 min). About 530 bp of the 25S rDNA D2-D3 region was amplified using the primers 25S4R (5′-ACAAGTGCTGAGTTCCTCAG-3′, a specific primer) and ITS4R (5′-GCATATCAATAAGCGGAGGA-3′ [Citation19], and PCR used the same thermocycle program as that for ITS. PCR products were prepared for sequencing using Antarctic Phosphatase and Exonuclease I (New England BioLabs, Australia) or the Macrogen purification service, then sequenced at Macrogen Inc. (Republic of Korea) using BigDye chemistry (Applied Biosystems) and the Sanger sequencing method using the same primers as that used for the initial amplification. All PCR products were sequenced in both the forward and reverse directions.
2.4. Phylogenetic analysis
Sequences were edited in GENEIOUS R9 [Citation20] and aligned using Multiple sequence Alignment based on Fast Fourier Transform (MAFFT v7.017) [Citation21] with options L-INS-I (iterative refinement method incorporating local pairwise alignments), gap opening penalty = 1.5, 200PAM/K = 2 scoring matrix, offset value = 0.123 for nucleotide sequences, then adjusted by eye in GENEIOUS so that indels were aligned.
For analysis of ITS, 75 sequences of fungus symbionts of fungus-gardening termite species of subfamily Macrotermitinae (representing 19 species and five genera) were included from Genbank, along with one related, non-gardened fungus species (Blastoporella zonata) as outgroup (Supplementary Material Table S1). For 25S, we used 63 sequences of the symbiotic fungus from Genbank. These sequences belong to 29 species and seven genera (). The non-gardened fungus species Arthromyces claviformis (EU708336) and Arthromyces matolae (EU708334) were included as outgroups. Although ITS and 25S sequences from Omani samples could have been concatenated because they represent a single functionally linked locus, this could not be done with Genbank data because the two DNA regions were sequenced from different studies using different specimens. This is also why the outgroups used for each DNA region were different.
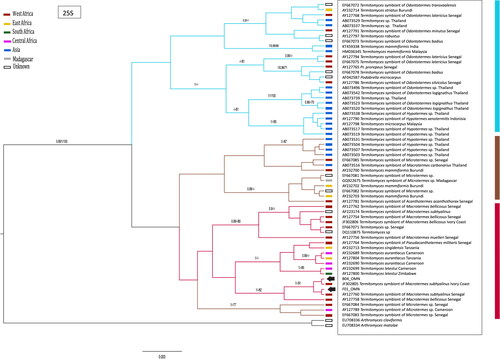
Figure 3. Phylogenetic relationships of the Omani populations of Termitomyces based on Bayesian analysis (BEAST) of the 25S gene. Support values are shown above nodes (MrBayes/BEAST/bootstrap support). Collection locality is indicated by colored bars next to tips. GenBank accession numbers correspond to those.
Identical sequences from Omani samples were collapsed into a single representative prior to phylogenetic analysis to decrease computational intensity and over-representation of alleles in model estimation. Both ITS and 25S were tested for deviation from stationarity [Citation22] using PAUP v4.0a146 [Citation23]. Phylogenetic analyses were performed using two methods (maximum parsimony and Bayesian inference) because each has differing underlying assumptions, and congruence across methods increases confidence in results. These were also compared with a neighbor-joining dendrogram [Citation24].
For neighbor-joining analyses, seeding was randomly initiated and the Hasegawa–Kishono–Yano (HKY) model [Citation25], with a gamma shape parameter of 0.5 was used to calculate the genetic distances among alleles as identified as the best-fit evolutionary model for both ITS and 25S genes by JMODELTEST [Citation26]. To evaluate support for nodes, fast stepwise addition bootstrapping using 1000 pseudoreplicates was used, and nodes with bootstrap values over 70% were regarded as supported (as per [Citation27].
Maximum parsimony searches were carried out using a heuristic search, 100 random addition replicates, tree bisection and reconnection (TBR) branch-swapping, all character positions weighted equally, gaps treated as missing data, and saving one tree only at each step. The shortest trees saved were then used as starting trees in a subsequent heuristic search using TBR branch swapping and saving all most-parsimonious trees. This approach was adopted to maximize the number of replicates performed on a large data set and to overcome becoming "stuck" on only a few "tree islands". A strict consensus tree was generated from these MP trees. Nodes were evaluated using 1000 bootstrap pseudoreplicates using the full heuristic search option and the settings as before.
In order to efficiently select the most appropriate model of sequence evolution for Bayesian analyses we used JMODELTEST [Citation26] and the Bayesian Information Criterion to select the best fitting model. This identified the Hasegawa–Kishono–Yano (HKY) [Citation25] including a proportion of invariable sites (I), and gamma-distributed among-site rate variation (Γ). Bayesian inference with an outgroup was used in MrBayes v3.2.6 [Citation28], sampling every 1000th generation of 10 million generations. Convergence of runs was assessed using Bayes Factors [Citation29] ensuring that the harmonic mean of the two runs was less than 2, and checking ESS values using TRACER v1.6 [Citation30]. Posterior probabilities ≥0.95 were considered as significant, as per [Citation31].
BEAST v1.8.2 [Citation32] was also used to estimate a phylogeny of Termitomyces alone because it does not require an outgroup. Two separate runs of 10 million generations were performed, sampling every 1000 generations. Runs were checked for stationarity as per MrBayes. Logcombiner was used to combine the results of the two runs, and TreeAnnotator was used to find the MCC (max clade credibility) tree. The analyses were run at least twice under both Yule and Birth-Death process model in tree priors to ascertain the effect of these tree priors on the resulting estimates [Citation33].
3. Results
There was no bias in base composition (non-stationarity) among alleles of each gene. Fungi from 61 of the 76 collection locations in Oman were successfully sequenced for ITS. All were identical and therefore only one representative sequence was used in further phylogenetic analyses. Termitomyces was generally grouped together based on their association with specific termite genera. However, there are some exceptions to this pattern. Some Termitomyces associated with thermite genera like Odontotermes, Microtermes, Pseudacanthotermes, and Cyptotrama formed polyphyletic groups. In contrast, Termitomyces associated with the genus Macrotermes formed a monophyletic group. Termitomyces associated with different Macrotermes species did not make monophyletic groups, but they formed several sub-lineages. For example, Termitomyces associated with Macrotemes bellicosus appeared in two lineages. One of them included Macrotemes bellicosus from Senegal and Kenya and the other lineage included Macrotemes bellicosus from Senegal, Macrotemes michaelseni from Kenya, Macrotemes herus from Kenya, and Macrotemes subhyalinus from Benin. Termitomyces associated with Macrotemes subhyalinus appeared in two lineages. Most of them were included in a monophyletic lineage majorly consisted of Macrotermes subhyalinus from Senegal, Benin, Ivory Coast, and Kenya and Macrotermes jeanneli from Kenya, and Macrotermes sp. from Kenya. ITS sequences from Termitomyces samples collected in Oman were identical and showed 3 bp difference with the Termitomyces associated with Macrotemes subhalinus from Benin. The Omani samples were nested within a monophyletic lineage consisting of Termitomyces associated with Macrotermes subhyalinus from West Africa. Separation of the lineage from the Termitomyces lineage associated with Macrotermes subhyalinus, Macrotermes jeanneli, and Macrotermes sp. from East Africa was weakly supported by bootstrap analysis. However, monophyly of them and sister relationship between the two lineages was maintained in maximum parsimony and Bayesian approaches.
The 25S gene was successfully amplified from 73 of the 76 fungal combs. There were multiple alleles present among Omani samples. Two alleles differed by a single nucleotide substitution, with 52 samples homozygous for one of these two alleles (45 for allele A, 7 for allele B) and 12 samples were heterozygous for alleles A and B. Another nine samples were polymorphic at two or more sites and alleles could not be phased. Alleles of 25S amplified from Termitomyces in Oman fell within a clade of symbionts of Macrotermes subhyalinus from Ivory Coast and Senegal, West Africa (): allele B was identical to sequences from Ivory Coast and Senegal, whereas allele A was sister to this clade.
4. Discussion
Despite the analyses of ITS and 25S comprising different samples and populations, the results were consistent.
Our finding that Omani samples of Termitomyces are most closely related to samples from Western Africa (Senegal, Benin, Ivory Coast), rather than collections from east Africa, was unexpected because Oman is geographically closer to east Africa. In addition, the vegetation in the Dhofar region of Oman, in which Termitomyces occurs, is part of the Somalia-Masai biome (also known as the Somalia-Masai regional center of endemism), which also spans parts of Eritrea, Ethiopia, Kenya, Somalia and Sudan in Eastern Africa. At least eight genera of plants in Oman are endemic to this biome [Citation18]. So, given the geographic proximity and similar climate and vegetation types, it could be expected that any dispersal between Africa and Oman would be between these areas. That a different pattern was found needs explanation.
In analyses of 25S, this unexpected biogeographic result could be an artifact of poor sampling from Africa. Alleles of 25S from Termitomyces from Western Africa are found across several clades in the phylogeny () but there have been very few samples from east Africa sequenced. If more extensive sampling was conducted, alleles more similar to those sampled from Oman might be uncovered. However, ITS has been better sampled from east Africa and the allele from Omani samples is clearly nested within a clade from West Africa (). Nevertheless, there are multiple clades in the phylogenies of both DNA regions that comprise samples from both Eastern and Western Africa, and sequences from Termitomyces associated with M. subhyalinus are also scattered across clades ( and )—there is clearly not a single east African clade and a single west African clade. Additional sampling of fungal combs across Africa may reveal new clades or many more examples of mixed clades, so it is not yet definitive that there are no Eastern African Termitomyces similar to those from Oman. An alternative explanation to insufficient sampling is that Termitomyces colonized Oman from Western Africa, either with the original colonization by Macrotermes subhyalinus or secondarily, replacing the original lineage of Termitomyces.
Figure 4. Phylogenetic relationships of the Omani populations of Termitomyces based on Bayesian analysis (BEAST) of the ITS region. Support values are shown above nodes (MrBayes/BEAST/bootstrap support). Collection locality is indicated by colored bars next to tips. GenBank accession numbers correspond to those in.
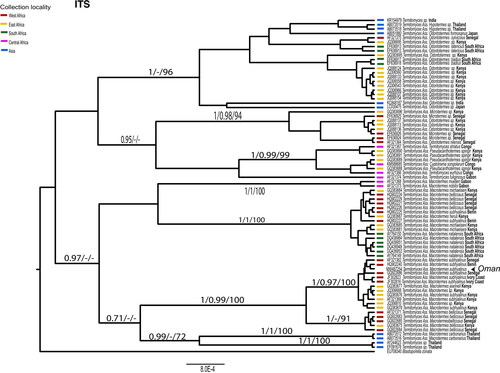
There is very little allelic diversity among the samples of Termitomyces from Oman, with the ITS region being identical across all samples sequenced. For both DNA regions (ITS and 25S), alleles sampled from Omani populations are nested among a diverse array of alleles sampled from Africa, indicating that Termitomyces in Oman is derived from African ancestry. That is, dispersal has occurred from Africa to Oman. The ITS allele and one allele of 25S from Oman were different from any sampled in Africa. If this differentiation is not due to insufficient sampling in Africa, it could indicate that Omani populations have been isolated for some time and evolved unique alleles. Nevertheless, the low diversity and nesting within clades of alleles from Africa indicate a single colonization event of Oman. More finely resolving genetic markers, such as SNPs derived from RADseq, are required to assess whether Omani populations show genetic structure consistent with long-term occupancy or whether they have only more recently arrived, potentially even within the human epoch.
5. Conclusion
Termitomyces symbionts of the fungus-gardening termites from Oman appear to share a more recent common ancestry with Termitomyces symbionts of M. subhyalinus from Western Africa than from Eastern Africa, but we cannot rule out alternative scenarios because of poor representation of other populations in Genbank. The phylogenetic pattern is consistent with dispersal to Oman from Africa. The lack of genetic diversity in Oman, and geographical contrasts in closest relative requires more investigation with more sampling from Termitomyces symbionts of M. subhyalinus is needed.
Author contributions
H. S. AlShamakhi was responsible for performing lab work and data analysis, interpreting the data, and drafting and writing the manuscript. L. G. Cook critically reviewed drafts and provided advice on analyses and interpretation of the data.
Supplemental Material
Download MS Word (19.7 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The datasets generated during and/or analyzed during the current study are available in the Supplementary Materials.
Additional information
Funding
References
- Batra LR, Batra SWT. Termite-fungus mutualism. In: Batra LR, editor. Insect-fungus symbiosis. Nutrition, mutualism and commensalism. New York (NY): Allanheld, Osmun and Co.; 1979. p. 117–163.
- Aanen DK, Boomsma JJ. Evolutionary dynamics of the mutualistic symbiosis between fungus-growing termites and Termitomyces fungi. In: Vega FE, Blackwell M, editors, Insect-Fungal Associations: ecology and Evolution. New York (NY): Oxford University Press; 2005. p. 191–210.
- Darlington JPEC. Nutrition and evolution in fungus-growing termites. In: Hunt JH, Nalepa CA, editors. Nourishment and evolution in insect societies. Boulder, CO: Westview Press; 1994. p. 105–130.
- Chapela IH, Rehner SA, Schultz TR, et al. Evolutionary history of the symbiosis between fungus-growing ants and their fungi. Science. 1994;266(5191):1691–1694.
- Mikheyev AS, Mueller UG, Abbot P. Comparative dating of attine ant and Lepiotaceous cultivar phylogenies reveals coevolutionary synchrony and discord. Am Nat. 2010;175(6):E126–E133. doi: 10.1086/652472.
- Mueller UG, Rehner SA, Schultz TR. The evolution of agriculture in ants. Science. 1998;281(5385):2034–2038. doi: 10.1126/science.281.5385.2034.
- Mueller UG, Schultz TR, Currie CR, et al. The origin of the attine ant-fungus mutualism. Q Rev Biol. 2001;76(2):169–197. doi: 10.1086/393867.
- Aanen DK, Eggleton P, Rouland-Lefevre C, et al. The evolution of fungus-growing termites and their mutualistic fungal symbionts. Proc Natl Acad Sci USA. 2002;99(23):14887–14892. doi: 10.1073/pnas.222313099.
- Donovan S, Jones D, Sands W, et al. Morphological phylogenetics of termites (Isoptera). Biol J Linnean Soc. 2000;70(3):467–513. doi: 10.1111/j.1095-8312.2000.tb01235.x.
- Kambhampati S, Eggleton P. Taxonomy and phylogenetics of Isoptera. In: Abe T, Bignell DA, Higashi M, editors. Termites: evolution, sociality, symbioses and ecology. Dordrecht: Kluwer Academic Publishers; 2000. p. 1–23. doi: 10.1007/978-94-017-3223-9_1.
- Miura T, Maekawa K, Kitade O, et al. Phylogenetic relationships among subfamilies in higher termites (Isoptera: termitidae) based on mitochondrial COII gene sequences. Ann Entomol Soc Am. 1998;91(5):515–523. doi: 10.1093/aesa/91.5.515.
- Rouland-Lefèvre C. Symbiosis with fungi. In: Abe T, Bignell DE, Higashi M, editors. Termites: evolution, sociality, symbioses, ecology. Dordrecht: Springer; 2000. doi: 10.1007/978-94-017-3223-9_14.
- Nobre T, Kone NA, Konate S, et al. Dating the fungus-growing termites’ mutualism shows a mixture between ancient codiversification and recent symbiont dispersal across divergent hosts. Mol Ecol. 2011b;20(12):2619–2627. doi: 10.1111/j.1365-294X.2011.05090.x.
- Rouland-Lefèvre C, Bignell DE. Cultivation of symbiotic fungi by termites of the subfamily Macrotermitinae. In: Seckbach J, editor. Symbiosis. Cellular origin, life in extreme habitats and astrobiology, vol 4. Dordrecht: Springer; 2001. p. 731–756. doi: 10.1007/0-306-48173-1_46.
- Nobre T, Fernandes C, Boomsma JJ, et al. Farming termites determine the genetic population structure of termitomyces fungal symbionts. Mol Ecol. 2011a;20(9):2023–2033. doi: 10.1111/j.1365-294X.2011.05064.x.
- Aanen DK, Ros VI, de Fine Licht HH, et al. Patterns of interaction specificity of fungus-growing termites and Termitomyces symbionts in South Africa. BMC Evol Biol. 2007;7(1):115. doi: 10.1186/1471-2148-7-115.
- Deng T, Zhou Y, Cheng M, et al. Synergistic activities of the symbiotic fungus Termitomyces albuminosus on the cellulase of Odontotermes formosanus (Isoptera: Termitidae). Sociobiology. 2008;51:733–740.
- Ghazanfar SA. Quantitative and biogeographic analysis of the flora of the sultanate of Oman. Global Ecol Biogeogr Lett. 1992;2(6):189–195. doi: 10.2307/2997660.
- White JS, Bruns T, Lee S, et al. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Shinsky JJ, et al., editors. PCR protocols: a guide to methods applications. Academic Press; 1990. p. 315–322.
- Kearse M, Moir R, Wilson A, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–1649. doi: 10.1093/bioinformatics/bts199.
- Katoh H, Miura T, Maekawa K, et al. Genetic variation of symbiotic fungi cultivated by the macrotermitine termite Odontotermes formosanus (Isoptera: Termitidae) in the Ryukyu archipelago. Mol Ecol. 2002;11(8):1565–1572. doi: 10.1046/j.1365-294x.2002.01535.x.
- Jermiin LS, Ho SY, Ababneh F, et al. The biasing effect of compositional heterogeneity on phylogenetic estimates may be underestimated. Syst Biol. 2004;53(4):638–643. doi: 10.1080/10635150490468648.
- Swofford DL. PAUP: Phylogenetic analysis using parsimony (and other methods), Version 4.0 Beta 10. Sunderland: Sinauer Associates; 2002.
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425.
- Hasegawa M, Kishino H, Yano T-A Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22(2):160–174. doi: 10.1007/BF02101694.
- Darriba D, Taboada GL, Doallo R, et al. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772–772. doi: 10.1038/nmeth.2109.
- Hillis DM, Bull JJ. An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Syst Biol. 1993;42(2):182–192. doi: 10.2307/2992540.
- Huelsenbeck JP, Ronquist F. MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics. 2001;17(8):754–755. doi: 10.1093/bioinformatics/17.8.754.
- Kass R, Raftery A. Bayes factors. J Am Stat Assoc. 1995;90(430):773–795. doi: 10.1080/01621459.1995.10476572.
- Rambaut A, Suchard M, Xie D, et al. Tracer v1.6; 2014. http://beast.bio.ed.ac.uk/Tracer
- Alfaro ME, Holder MT. The posterior and the prior in Bayesian phylogenetics. Annu Rev Ecol Evol Syst. 2006;37(1):19–42. doi: 10.1146/annurev.ecolsys.37.091305.110021.
- Drummond AJ, Suchard MA, Xie D, et al. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29(8):1969–1973. doi: 10.1093/molbev/mss075.
- Gernhard T. New analytic results for speciation times in neutral models. Bull Math Biol. 2008;70(4):1082–1097. doi: 10.1007/s11538-007-9291-0.