
Abstract
Lung cancer is a serious health problem, since it is one of the leading causes for death worldwide. Molecular–cytogenetic studies could provide reliable data about genetic alterations which could be related to disease pathogenesis and be used for better prognosis and treatment strategies. We performed whole genome oligonucleotide microarray-based comparative genomic hybridization in 10 samples of non-small cell lung cancer. Trisomies were discovered for chromosomes 1, 13, 18 and 20. Chromosome arms 5p, 7p, 11q, 20q and Хq were affected by genetic gains, and 1p, 5q, 10q and 15q, by genetic losses. Microstructural (<5 Mbp) genomic aberrations were revealed: gains in regions 7p (containing the epidermal growth factor receptor gene) and 12p (containing KRAS) and losses in 3p26 and 4q34. Based on high amplitude of alterations and small overlapping regions, new potential oncogenes may be suggested: NBPF4 (1p13.3); ETV1, AGR3 and TSPAN13 (7p21.3-7p21.1); SOX5 and FGFR1OP2 (12p12.1-12p11.22); GPC6 (13q32.1). Significant genetic losses were assumed to contain potential tumour-suppressor genes: DPYD (1p21.3); CLDN22, CLDN24, ING2, CASP3, SORBS2 (4q34.2-q35.1); DEFB (8p23.1). Our results complement the picture of genomic characterization of non-small cell lung cancer.
Introduction
Lung cancer is a leading cause of cancer-related death all over the world. It is classified into two types: small-cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC).[Citation1] NSCLC is more common and makes up about 85% of all lung cancers.[Citation2] Although the majority of NSCLC is caused or induced by cigarette smoking, 10% of the patients are non-smokers.[Citation3] Lung carcinogenesis – like the development of other cancers – is a multistage process and involves alterations in multiple genes and diverse pathways. Inherited polymorphisms in a number of genes, especially in carcinogen-metabolising genes, affect the individual susceptibility to development of lung cancer.[Citation4,5] More frequent changes include chromosomal rearrangements, microsatellite instability, deregulated expression of telomerase and alterations in angiogenesis. Mutational activation of oncogenes and inactivation of tumour-suppressor genes, and subsequent increased genetic instability are major genetic events in lung carcinogenesis (reviewed in [Citation6–8]). By the time lung cancer is clinically diagnosed, as many as 10–20 genetic alterations may have accumulated.[Citation9]
High-resolution microarray-based comparative genomic hybridization (array CGH) is a modern molecular technology used for performing a complete scan of genomic DNA. It demonstrates the presence of genetic losses and/or increased copy numbers of the genetic material in the tumour tissue and is characterized by high sensitivity and reliability of the results. The method is widely used for screening genomic analysis in a large number of malignancies.
In this study, we performed whole genome microarray analysis for screening of copy number changes in NSCLC in order to assess the genomic instability in different stages of this tumour type.
Subjects and methods
Subjects
In the microarray study, we used DNA isolated from tumours of 10 patients diagnosed with primary NSCLC. The clinical characteristics of the patients are summarized in . Six patients were male and four, female. The average age of the patients was 61 years (ranging from 52 to 67 years).
Table 1. Clinical data about the analysed lung cancer cases.
Materials
The materials were taken after tumour resection in the Department of Thoracic Surgery ‘St. Sofia’ during the period November 2007–December 2009 and were kept in the tissue bank of the Department of Medical Genetics. The collection of samples was approved by the Institutional Ethics Committee of the Medical University of Sofia and all participants signed informed consent forms. All tumours were staged postoperatively according to the classification system of the International Union Against Cancer.[Citation10] Among the 10 tumours with NSCLC, five had the histology of adenocarcinoma (AC) and five, of squamous cell carcinoma (SCC).
DNA extraction
Total DNA was extracted from the tissue samples, using QIAamp DNA minikit (Qiagen, Hilden, Germany). DNA concentration was measured spectrophotometrically by using a NanoDrop® ND-2000 spectrophotometer working with volumes of 1–2 μL. The 260/280 ratio was in the range of 1.8–2.0 for each sample. As an additional quality control, DNA was checked in a 1% agarose gel: DNA of high molecular weight (>50 kbp) indicated it suitable for use.
Comparative genomic hybridization on DNA microarrays
The principle of microarray-based comparative genomic hybridization is based on competitive hybridization between alternatively labelled tumour and normal DNA on slides with spotted DNA sequences (oligonucleotides or bacterial artificial chromosomes (BAC)). We used genomic array CytoChip (BlueGnome, Cambridge, UK) Oligonucleotide microchips 2×105 K with a density of 105.072 sequences covering the entire genome with a resolution of 35 Kb. CytoChip Oligo microchips have two separate fields for hybridization, allowing for simultaneous analysis of two samples on one chip. Each chip has a unique code at the bottom of the glass.
Array CGH probe labelling, hybridization, image capture and data analysis
The control and reference DNA was labelled by random priming, using Blue Gnome Fluorescent Labelling System. The labelled products were purified by AutoSeqTM G50 columns. The labelling mix was added in each tube containing DNA and primers; test DNA was labelled by fluorochrome Cy3 and control DNA, by Cy5. Hybridization was done by dissolving precipitated probes in hybridization buffer. Arrays were washed in standard saline citrate solutions with decreasing concentrations and scanned by a GenPix 4100A. All data were processed with the program BlueFuse Multi version 2.2 (BlueGnome, Cambridge). In data processing, base 2 logarithm (log2) ratios of Cy3 and Cy5 intensities are generated for all hybridized clones. Normal copy numbers are considered to be in the range of −0.3 to +0.3; values above +0.3 were evaluated as gain/amplification and those under −0.3, as losses (deletions). Genomic profiles were represented with logarithmic ratios on the Y-axis and along the 23 chromosomes on the X-axis. Individual chromosomal profiles were represented with clone positions on the Y-axis and logarithmic ratios on the X-axis.
Statistical analysis
Contingency table analysis and χ2 test were used to assess the relationship between gene copy number changes and tumour phenotype, i.e. tumour stage. P < 0.05 was considered as statistically significant.
Results and discussion
Array CGH and copy number aberrations in lung cancer
Lung cancer is a serious health problem because it is one of the leading causes of cancer mortality worldwide. To the best of our knowledge, we report for the first time the results from whole genome array CGH analysis in Bulgarian patients diagnosed with primary NSCLC. Array CGH is the most powerful tool for genetic screening of tumours.[Citation11,12] Its ability to simultaneously detect DNA copy number changes at multiple loci over the whole genome and to provide high-resolution mapping of variation in copy numbers was used in our study. Candidate genes responsible for disease can be identified; thus, the results could lead to new discoveries or could confirm the current data.[Citation13] They could help in the better understanding of the mechanisms of the disease by revealing potential oncogenes and tumour-suppressor genes located in aberrant regions revealed in our patients.
Our array CGH results showed that the average number of pathological aberrations per tumour was 10.1, among which genetic losses were prevalent. The average copy number loss per tumour was 5.8 and the average copy number gain per tumour was 4.3. The most frequent aberrations detected in our study were genetic gains of 7p (containing the epidermal growth factor receptor gene EGFR) and 12p (containing KRAS) and genetic losses of 3p26 and 4q34. Genetic losses were more frequent than gains in our study. In other studies, there are different data: some report prevalence of genetic losses,[Citation12,Citation14] while others, of genetic gains.[Citation15,16]
Analysis of large and regional (greater than 5 Mbp) aberrations
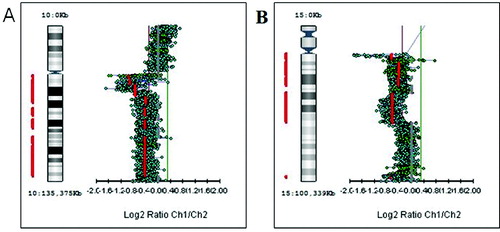
First, we studied large aberrations involving whole chromosomes or chromosome arms. We found in different tumours additional copies of whole chromosomes (trisomies) for chromosomes 1, 13, 18 and 20 (+1, +13, +18 and +20) as shown in . There were genetic gains for the following chromosome arms: the short arm of chromosome 5 (5p +), the short arm of chromosome 7 (7p+), the long arms of chromosomes 11 (11q+), X (Xq+), 14 (14q+) and 20 (20q+) in different tumours (). There were genetic losses for the following chromosome arms: the short arm of chromosome 1 (1p−), the long arms of chromosomes 5 (5q−), 10 (10q−) and 15 (15q−) in different tumours (). All these large aberrations were found in the group of early carcinomas.
Figure 1. Large aberrations such as gains of whole chromosomes (trisomies): trisomy 1 (A), trisomy 13 (B), trisomy 18 (C) and trisomy 20 (D).
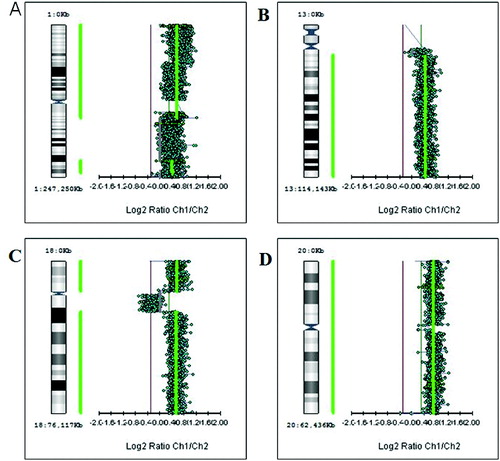
Figure 2. Genetic aberrations involving chromosome arms. (A) Gain of the short arm of chromosome 5 (5p+). (B) Gain of the long arm of chromosome 11 (11q+). (C) Gain of the long arm of chromosome 20 (20q+). (D) Gain of the long arm of X chromosome (Xq+).
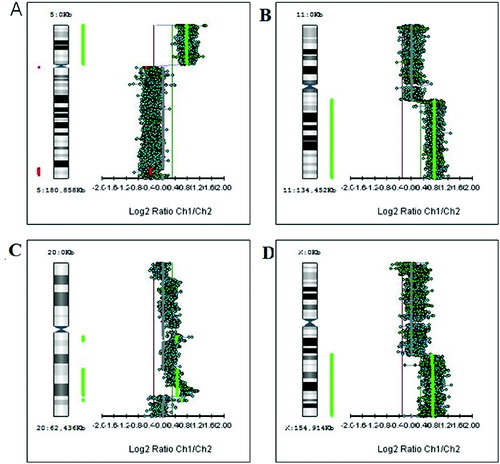
Figure 3. Genetic losses of chromosome arms. (A) Deletion of the long arm of chromosome 10 (10q−). (B) Deletion of the long arm of chromosome 15 (15q−).
The second step in our study was to analyse the so-called regional aberrations, which are larger than 5 Mbp. We observed regional genetic gains in chromosomes 4, 6, 7, 10, 11, 12, 13, 16 and X (). The highest frequency was revealed for regional gains of 7p21.3-p21.1 and 12p12.1-p11.22 – in 20% of the tumours. Regional genetic losses were found in chromosomes 1, 2, 3, 4, 5, 6, 7, 9, 11, 12, 13, 14, 15, 16, 17, 18, 19 and X (). The highest frequency was detected for regional losses of 3p26.2-p26.1 and 4q34.2-q35.1 – in 20% of the tumours.
Table 2. Regional genetic gains.
Table 3. Regional genetic losses.
In the group of early stage cancers, the most common types of aberrations were large ones (average of two large aberrations per tumour) and regional aberrations (average 5.6 regional aberrations per tumour) as compared with advanced cancers, where we observed zero large aberrations per tumour and zero regional aberrations per tumour (p < 0.03). Losses of whole chromosomes or chromosome arms are found in early stages of carcinogenesis and contribute to overall genomic instability of tumours. One of the first and earliest signs of lung epithelium in NSCLC is exactly the loss of the short arm of chromosome 3,[Citation17] which was also found in two of the tumours in our study, both from stage IB.
Another common genetic aberration characteristic of early lung carcinogenesis is deletion of chromosome 9.[Citation17] In our study, we found loss of the short arm of chromosome 9 in a tumour from stage IB. We also observed large aberrations involving whole chromosomes (+1, +13, +18 and +20) or chromosome arms (1p−, 5p+, 5q−, 7p+, 10q−, 11q+, 14q+, 15q−, 20q+ and Xq+). There was also regional genetic loss in 1p21.3-p13.1 and high amplitude loss in the same region.
Deletions of the short arm of chromosome 1 are common among various cancers. Nomoto et al. [Citation18] identified common unbalanced changes in 1p36 in breast cancer. This is the chromosomal region where the tumour-suppressor gene TP73, which shows significant homology with the TP53 gene, is located. Liu et al. [Citation3] examined the TP73 gene in six NSCLC cell lines and found abnormal methylation in exon 1 and loss of expression at mRNA and protein level. The change in methylation of TP73 may play an important role in the mechanism of silencing gene expression as well.[Citation3] In our experiments, among patients with early stage NSCLC, early genetic changes affecting 5p were identified. Here, we detected gain of 5p15.33. This locus harbours genes TERT, SLC6A19 and SLC6A18.
Analysis of microstructural aberrations (less than 5 Mb)
We selected the aberrations that have a high amplitude (log2 ratio T/N > 0.5 for genetic gains and <−0.5 for genetic losses). Following this approach, 42 aberrations were selected. Of these, 18 were genetic gains () and 24, genetic losses (). There was amplification of the same locus, 1q31.3, in two of the analysed tumours (). Also in two other tumours, there was loss of a single region, 8p23.1 ().
Table 4. Genetic gains with size <5 Mbp and high amplitude (log2 ratio T/N > 0.5) .
Table 5. Genetic losses with a size of <5 Mbp and high amplitude (log2 ratio T/N < −0.5) .
Microstructural aberrations were significantly more common in the group of advanced cancers: seven microstructural aberrations per tumour in late-stage tumours, as compared to four microstructural aberrations per tumour in early stage cancers (p < 0.006).
Potential candidate oncogenes from regions with copy number changes could include: CEP72, TPPP, AHRR, EXOC3, SLC9A3, LOC442126, ZDHHC11, BRD9, TRIP13, CLPTM1L, SLC6A3 and LOC401169. In the analysis of genomic regions with small aberrations, the most common types of aberrations were genetic gains with known role in tumourigenesis: in 7p (containing the oncogene EGFR) and 12p (containing the oncogene KRAS). We could suggest some new possible candidate oncogenes based on high-amplitude amplification and/or location in the small regions of overlap: NBPF4 (1p13.3); ETV1, AGR3 and TSPAN13 (7p21.3-7p21.1); SOX5 and FGFR1OP2 (12p12.1-12p11.22); GPC6 (13q32.1). There were regions of significant genetic losses that may prove useful in identifying potential tumour suppressor genes as possible candidates: DPYD (1p21.3); CLDN22, CLDN24, ING2, CASP3, SORBS2 (4q34.2-q35.1); DEFB (8p23.1).
Although further studies with a larger sample size would be needed to verify these speculations, our results contribute to the knowledge about the genomic aspects of NSCLC. There has been a great progress in understanding the complex mechanisms of tumourigenesis. Different genetic alterations suggest differences in clinical behaviour and therapeutic response of different tumour subtypes. Owing to the ever increasing opportunities that are open in the genomic era, researchers are able to discover new target therapies specific to each subtype of cancer, and even individual therapy according to the genomic profile of each patient. Molecular profiling of tumours is an important approach in determining prognosis and identifying patients who may respond well to specific therapy. The application of comparative genomic hybridization on DNA microarrays with high resolution allows the establishment of such specificity at chromosome and genetic level, which could help the clinical management of the patients.
Conclusions
This study is, to the best of our knowledge, the first report on whole genome array CGH analysis in Bulgarian patients diagnosed with primary NSCLC. Comparative genomic hybridization on DNA microarrays with high resolution allows some tumour specifics to be observed at chromosome and genetic level, which could help in the clinical management of the patients. Our results suggested that early stage lung cancers are characterized by large chromosomal aberrations, whereas late-stage tumours harbour microstructural aberrations containing gene amplifications or deletions. Their expression levels are worthy of being investigated as a step towards discovery of new biomarkers. Further studies with a larger sample size would be needed to verify these speculations.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Brambilla E, Travis WD, Colby TV, Corrin B, Shimosato Y. The new World Health Organization classification of lung tumours. Eur Respir J. 2001;18(6):1059–1068.
- American Cancer Society [Internet]. American Cancer Society, Inc.; c2014 [cited 2014 Oct 10]. Available from: http://www.cancer.org/cancer/lungcancer-non-smallcell/.
- Liu K, Zhan M, Zheng P. Loss of p73 expression in six non-small cell lung cancer cell lines is associated with 5’CpG island methylation. Exp Mol Pathol. 2008;84(1):59–63.
- Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83(5):584–594.
- Leelakumari S, Madhavan J, Ankathil R. A multigenic approach to predict risk and survival among Indian lung cancer patients: P1-042. J Thorac Oncol. 2007;2(8):S566.
- Cherneva R, Dimova I. Genetic aspects of lung cancer. Balkan J Med Genet. 2006;9(1–2):9–17.
- Al Zeyadi MM. Molecular and genetic aspects of lung cancer. Biotechnol Biotechnol Equipment. 2013;27(5):4051–4060.
- Herbst RS, Heymach JV, Lippman SM. Molecular origins of cancer: lung cancer. New England J Med. 2008;359:1367–1380.
- Haigentz M, Perez-Soler R. Chemopreventive therapeutics inhalation therapies for lung cancer and bronchial premalignancy. Methods Mol Med. 2003;75:771–780.
- International Union Against Cancer [Internet]. TNM classification of malignant tumours. UICC; c2014 [cited 2014 Oct 10]. Available from: http://www.uicc.org/resources/tnm.
- Albertson DG, Ylstra B, Segraves R, Collins C, Dairkee SH, Kowbel D, Kuo WL, Gray JW, Pinkel D. Quantitative mapping of amplicon structure by array CGH identifies CYP24 as a candidate oncogene. Nat Genet. 2000;25(2):144–146.
- Dehan E, Ben-Dor A, Liao W, Lipson D, Frimer H, Rienstein S, Simansky D, Krupsky M, Yaron P, Friedman E, Rechavi G, Perlman M, Aviram-Goldring A, Izraeli S, Bittner M, Yakhini Z, Kaminski N. Chromosomal aberrations and gene expression profiles in non-small cell lung cancer. Lung Cancer. 2007;56(2):175–184.
- Brena RM. Aberrant DNA methylation in human non-small cell lung cancer [ dissertation]. Columbus (OH): The Ohio State University; 2007. Available from: https://etd.ohiolink.edu/.
- Boelens MC, Kok K, van der Vlies P, van der Vries G, Sietsma H, Timens W, Postma DS, Groen HJ, van den Berg A. Genomic aberrations in squamous cell lung carcinoma related to lymph node or distant metastasis. Lung Cancer. 2009;66(3):372–378.
- Baik SH, Jee BK, Choi JS, Yoon HK, Lee KH, Kim YH, Lim Y. DNA profiling by array comparative genomic hybridization (CGH) of peripheral blood mononuclear cells (PBMC) and tumor tissue cell in non-small cell lung cancer (NSCLC). Mol Biol Rep. 2009;36(7):1767–1778.
- Sung JS, Park KH, Kim YH. Genomic alterations of chromosome region 11p as predictive marker by array comparative genomic hybridization in lung adenocarcinoma patients. Cancer Genet Cytogenet. 2010;198(1):27–34.
- Wistuba II, Behrens C, Virmani AK, Mele G, Milchgrub S, Girard L, Fondon JW 3rd, Garner HR, McKay B, Latif F, Lerman MI, Lam S, Gazdar AF, Minna JD. High resolution chromosome 3p allelotyping of human lung cancer and preneoplastic/preinvasive bronchial epithelium reveals multiple, discontinuous sites of 3p allele loss and three regions of frequent breakpoints. Cancer Res. 2000;60(7):1949–1960.
- Nomoto S, Haruki N, Tatematsu Y, Konishi H, Mitsudomi T, Takahashi T, Takahashi T. Frequent allelic imbalance suggests involvement of a tumor suppressor gene at 1p36 in the pathogenesis of human lung cancers. Genes Chromosomes Cancer. 2000;28(3):342–346.