
Abstract
Cellulase is a widely used enzyme for saccharification of plant cell walls for the production of biofuels. In our work, we tested an ethanol inducible native alcA promoter from Aspergillus nidulans in Escherichia coli (E. coli) and tobacco. The alcA promoter was fused with a cellulase gene and then the promoter was compared with other widely used promoters, namely, T5 and cauliflower mosaic virus (CaMV) 35S in E. coli. We also tested the activity of alcA promoter in tobacco by transiently expressing a green fluorescent protein gene under the control of alcA promoter. We concluded from our quantitative polymerase chain reaction (qPCR) results that when alcA promoter was expressed along with AlcR transcription factor, the alcA promoter was 11 times more active than the T5 promoter. The qPCR data suggested that there may be a direct correlation between AlcR concentrations and alcA activity, indicating a key role of the activator AlcR in the regulation of the alcA promoter. Our work on inducible promoter system for cellulase production may open new avenues of research in the field of biofuel development from living cells.
Introduction
The recent rise in oil prices, along with the growing concern about global warming, has prompted scientists to delve deeper into the field of alternative energy research. Currently, bioethanol and biodiesel are the two biofuels that present the most interesting options. Bioethanol production can be achieved through bioconversion of lignocellulosic materials. Such process is high mass fermentation of sugar present in plant cell walls. However, the strong associations between the long chains of polysaccharides in plant cell walls are a challenge, which requires harsh conditions, in order to break down the recalcitrant cell wall.[Citation1,Citation2] Although addition of lignocellulosic degrading enzymes (including cellulases, hemicellulases and pectinases) have been found useful for the conversion of biomass into fermentable monomeric sugars, their large-scale applications are costly.[Citation3] An optimization of the enzymatic degradation of lignocellulose is required in order to develop an economically feasible process for bioconversion of lignocellulosic biomass to ethanol. Genetically modified micro-organisms for the bioconversion of lignocellulosic materials have recently been demonstrated by several studies.[Citation3–5] Many of these studies have focused on the engineering of Escherichia coli (E. coli) for bioethanol production, because of E. coli's high amenability to metabolic modifications, as well as its ability to ferment a wide variety of sugars into a broad range of short-chain alcohols.[Citation6–9] Moreover, it has been reported that processes conducting simultaneous cellulose hydrolysis by cellulase enzyme and fermentation lead to a higher ethanol production and lower enzyme consumption.[Citation10] However, this method needs compatible pH and temperature conditions for both enzyme producing and ethanologenic micro-organisms.[Citation10] Developing bacterial co-cultures with genetically engineered micro-organisms capable of conducting the entire task from start to finish would solve the compatibility problem and would lead to a significant cost reduction in the ethanol production procedure.
One of the strongest inducible gene expression systems in the filamentous fungus Aspergillus nidulans (A. nidulans) is the alc regulon, which encodes for the ethanol utilization pathway.[Citation11] The ethanol utilization pathway in A. nidulans is a highly inducible regulatory system for the consumption of ethanol as a carbon source. The pathway requires a specific activator (AlcR) (encoded by alcR gene) mediating the transcription, in the presence of an inducing compound (e.g. ethanol).[Citation12] The alcR-alcA genes, the main elements of the alc regulon, are widely used for the high-level expression of heterologous proteins.[Citation13,Citation14] This system has been shown to have low basal activity and high induction level of gene expression in plants.[Citation15] It is composed of two elements – the one is alcA promoter, which regulates the expression of alcohol dehydrogenase I and the other one is alcR gene, which encodes for the transcription activator (AlcR) mediating the transcription induction in the presence of an inducing compound.[Citation11,Citation16] The activator protein AlcR responds to the inducer molecule (ethanol, ethyl methyl ketone, etc.) and binds to specific regions within the alcA promoter.[Citation11,Citation15,Citation17] This system is useful for transgene expression, since it is inexpensive, effective for dose-dependent expression and suitable for use both in laboratory and in field.[Citation14]
The efficiency of alc regulon has frequently been studied in fungi and plants, but to the best of our knowledge, there have not been any reports of such studies in E. coli. Here, we characterized in detail the efficacy of alc system, in comparison with two other promoters, for their suitability to express foreign genes in E. coli and Nicotiana tabacum. Three potentially useful promoters have been included in this comparison: native alcA promoter from A. nidulans, T5 promoter from coliphage and 35S promoter from cauliflower mosaic virus. All these three promoters were used to drive the endo-1,4-beta-D-glucanase (Umcel9B) [Citation18] gene expression in E. coli. The 35S and native alcA promoters were used to drive green fluorescent protein (GFP) gene expression in tobacco (N. tabacum). The Umcel9B gene (referred as cellulase herein) which belongs to the glycoside hydrolase family 9 (GHF 9), is a novel cellulase enzyme identified from metagenome of the compost soil. It has been reported that recombinant protein of cellulase has an endoactive enzyme activity in E. coli.[Citation18] This cellulase gene was expressed in E. coli under the control of various promoters to accurately analyse the in vivo strength of these three well-defined promoters.
Materials and methods
Bacterial strains and expression vectors
E. coli DH5α and One Shot TOP10 (Invitrogen, USA) strains were used as expression hosts and cultured in Luria-Bertani (LB) medium. TOP10 strain, which is similar to DH10B strain, was used as a host for pT5 (empty) and pT5-cel (cellulase is abbreviated as ‘cel’ throughout the paper) constructs due to incompatibility of pQE-30 vector with DH5α strain. The cellulase gene (DQ235086) [Citation18] was synthesized by GenScript, USA, in pUC57 vector with BamHI and SacI restriction sites at the ends. The gene was used to construct four vectors by using three different types of carriers – pBIN mgfp5ER,[Citation19] pQE-30 Xa (Qiagen, USA) and pCAMBIA1200 (Marker Gene, USA). The pBIN mgfp5ER originally contains a constitutive promoter cauliflower mosaic virus (CaMV 35S) and gfp as a reporter gene. The pUC57 vector was digested with BamHI and SacI releasing the cellulase gene, which was inserted into the pBIN and pQE (pQE carries T5 promoter) vectors. The following cassettes were made through this process – p35S-cel and pT5-cel. The p35S-cel vector was further treated with HindIII and XbaI to remove the 35S promoter and replace it with alcA promoter (generously provided by Syngenta, USA) to make palcA-cel cassette. The palcA-gfp construct was also made by using p35S-gfp (pBIN mgfp5ER). The alcR gene (Syngenta, USA) was digested with EcoRI and HindIII and cloned into the pCAMBIA vector to make p35S-alcR cassette. palcA-cel-alcR refers to bacterial cells harboring both palcA-cel and p35S-alcR plasmids. The presence of genes in all constructs was confirmed by sequencing (Laragen, USA). The details of the vector constructions are shown in . All bacterial strains, plasmids and primers used in this paper are listed in
Figure 1. Plasmid constructs used in this study.
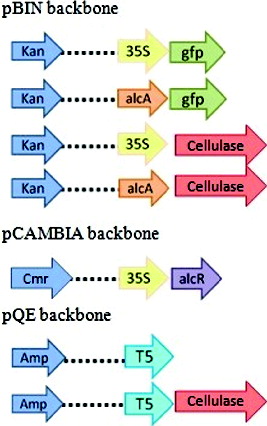
Table 1. Bacterial/plant strains, plasmids and primers used in this study.
Ethanol induction conditions
Both E. coli and Agrobacterium strains harboring alcA promoter were tested for ethanol toxicity in liquid medium. E. coli cells were grown in LB medium at 37 °C for 24 h and Agrobacterium cells in yeast extract broth (YEB) media (10 g/L peptone, 10 g/L yeast extract, 5 g/L NaCl) at 28 °C for two days with various ethanol concentrations (0%–5% (v/v) with 1% increment). Liquid cultures were incubated in a shaking incubator at an agitation of 200 rpm. Cell viability assay was performed for E. coli cells grown on agar plates containing different ethanol concentrations. DH5α cells were grown at 37 °C overnight and then the cultures were serially diluted up to 10−7. A quantity of 100 µL of 10−6 and 10−7 diluted cultures was spread on LB plates with different ethanol concentrations ranging from 0% to 8% (v/v) with 1% increment. The colonies were counted after an overnight incubation at 37 °C. Three technical replications were conducted for each dilution per each ethanol concentration. The 0% ethanol was used as negative control.
Total RNA extraction and cDNA synthesis
Total RNA was extracted from E. coli cells by using Quick-RNA MiniPrep Kit (Zymo Research, USA) according to the manufacturer's instructions. The RNA was then treated with DNase I and Turbo DNase (Life technologies, USA) several times to remove the genomic contamination completely and then was purified by using RNA Clean-Up kit (Zymo Research, USA). Complementary DNA (cDNA) was synthesized from 1 µg total RNA extracted from overnight grown E. coli cell culture by using iScript cDNA Synthesis Kit (Bio-Rad, USA), according to the manufacturer's instructions. cDNAs of more than three biological samples were prepared as mentioned above and stored in −20 °C. Genomic DNA contamination was checked visually using gel electrophoresis (1% agarose) after reverse transcription polymerase chain reaction (RT-PCR). RT-PCR was also used to check the absence or presence of cellulase gene expressions in p35S-cel, pT5-cel, palcA-cel, p35S-gfp, pT5 (empty) and palcA-cel-alcR strains, alcR gene expression in palcA-cel-alcR and palcA-gfp-alcR strains and gfp gene expression in palcA-gfp and palcA-gfp-alcR strains. The transcription of the three genes was targeted by correlating detection primers (). RT-PCR was conducted by using 0.3 µL and 0.5 µL cDNA, 1 µL 10 µmol/L of each of the forward and reverse primers and PCR Master Mix (Fermentas, USA). PCR negative controls (C−) (containing primers and PCR Master Mix) and no reverse transcriptase (NRT) controls (containing RNA, primers and PCR Master Mix) were included for each RT-PCR test to check for any DNA contamination. PCR was performed at an initial denaturation at 95 °C for 2 min, 40 cycles with initial denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s and extension at 72 °C for 2 min, with a final extension at 72 °C for 15 min. p35S-gfp and pT5 (empty) strains were used as negative controls to check the transcriptional expression of cellulase gene.
Quantitative real-time PCR
Quantitative real-time PCR (qPCR) reaction was conducted using iTaq Universal SYBR Green Supermix (Bio-Rad, USA) in qPCR tubes. The cysG (cysteine) gene was used as a reference gene for E. coli in this study. The reaction mixture consisted of 1 μL cDNA samples, 1 μL of each of the forward and reverse primers (2 μmol/L) (), 5μL iTaq Universal SYBR Green Supermix (2 ×) and 2 μL nuclease free water in a final volume of 10 μL. Three technical replicates were performed for each biological sample, reference gene, NRT and PCR negative control (C−). The qPCR protocol was as follows – 30 s initial denaturation at 95 °C, 39 cycles of denaturation at 95 °C for 5 s and annealing at 60 °C for 30 s, followed by a melting curve analysis at 65–95 °C with 0.5 °C increment for 2–5 s per step. Melting curve analysis was performed to verify the specificity of each primer. The threshold cycle (Ct) values were calculated for each reaction by the CFX96 Touch Real-Time PCR (Bio-Rad, USA) set with default parameters. The normalized relative expression values (ΔΔCt) were analysed relative to zero by Bio-Rad CFX Manager 3.1. The ΔΔCt values of various promoters are mentioned in . Transcription of cellulase and alcR genes was studied by qPCR to evaluate their expression levels under the control of an upstream promoter.
Table 2. The magnitude of fold differences between all upregulated samples.
Transient expression experiment in tobacco
Transient expression assay was performed using a native alcA promoter from A. nidulans in tobacco. By this method, the time course of induction and gene expression of alcA, compared with 35S promoter, was characterized. Tobacco (Nicotiana tabacum L. cv. Petit Havana), used for transient expression experiment, was kindly provided by Dr Ernest Kwok, California State University, Northridge. The palcA-gfp and p35S-gfp were transferred into Agrobacterium tumefaciens strain LB4404 through electroporation. Agrobacterium cells were transformed using standard electroporation protocol.[Citation20] Cells containing palcA-gfp cassette were further used for transferring p35S-alcR cassette, in which one cell contains both vectors. Agrobacteria were then grown in 5 mL YEB medium (10 g/L peptone, 10 g/L yeast extract, 5 g/L NaCl) supplemented with antibiotics: rifampicin (100 µg/mL), kanamycin (50 µg/mL) and chloramphenicol (35 µg/mL) at 28 °C for 2 days. Bacterial suspension was adjusted to the OD600 of 0.8. Cell culture (1 mL) was pelleted by centrifugation for 15 min at 3000 g. Then, cells were resuspended in 1.2 mL of 10 mmol/L 2-(N-morpholino) ethanesulfonic acid (MES, PH 5.5) plus Murashige and Skoog (MS) basal medium [Citation21] and supplemented with 100 µmol/L acetosyringone.[Citation22] Agroinfiltration was conducted on a fully expanded mature leaves by using a 1 mL plastic syringe. Each spot (about 3–4 cm2 surrounding the infiltrated area) was targeted by infiltrating 100 µL of bacterial suspension. Some leaves treated with cells containing alcA promoter were immediately infiltrated with 2% ethanol solution after cells infiltration while others were kept uninduced, as a control. Fluorescent activity of these lines was compared to wild-type leaves. The infiltrated areas were marked and the plants were maintained in a green house at 22 °C.
Epifluorescence microscopy
Several 3-mm wide sections of marked areas on the tobacco leaves were cut and mounted on the slide. Images were generated using confocal laser scanning microscope (Zeiss Observer.Z1 microscope) with 40 × objective lens. Fluorescence visualization in tobacco leaves was monitored every 24 h for 10 days after infiltration. Fluorescent visualization of samples was inspected using differential interference contrast and GFP fluorescence images, as well as their overlap.
Statistical analysis
The transcriptional analysis by real-time PCR was investigated by three independent trials in triplicate for each trial. Data were analysed using one-way analysis of variance with Bonferroni Multiple Comparisons Test and Tukey-Kramer Multiple Comparisons Test. The test was performed using GraphPad InStat version 3.0a for Macintosh (GraphPad Software, USA).
Results and discussion
Ethanol induction conditions
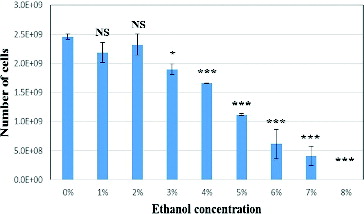
Ethanol induction with various concentrations was performed for both E. coli and Agrobacterium strains harbouring alcA promoter to check for ethanol toxicity in the liquid medium. Both bacterial strains generated the same results (data for Agrobacterium are not shown). Cell growth was observed in the overnight liquid cultures with 1% and 2% ethanol with OD600 above 2.5. There was no cell growth in the other ethanol concentrations. Cell viability test for E. coli cultures grown on agar plates with 0% to 7% ethanol exhibited growth of colonies, although significant decrease in growth rate was observed at higher ethanol concentrations (). Optimum growth was obtained on agar plates with 1% and 2% ethanol concentrations. According to our data and another study,[Citation23] considering both liquid and solid cultures, the lowest ethanol toxicity associating optimum growth can be obtained using 1% and 2% ethanol concentrations which were used for further experiments.
Figure 2. Ethanol toxicity assay for E. coli cultures.
Analysis of cellulase gene expression regulated by different promoters
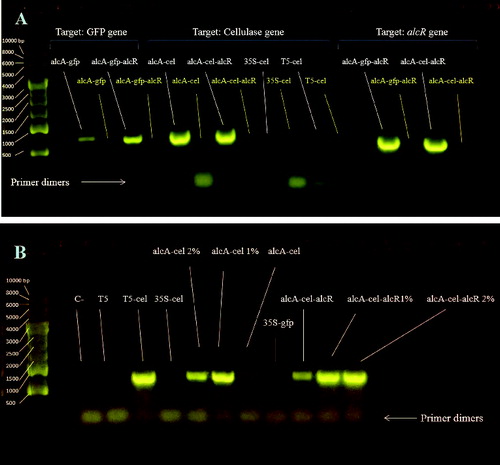
We conducted a preliminary RT-PCR test to check the absence or presence of cellulase gene expressions in the following constructs: p35S-cel, pT5-cel, palcA-cel, p35S-gfp, pT5 (empty) and palcA-cel-alcR. Gel electrophoresis data revealed bands for all samples, except p35S-cel, p35S-gfp and pT5 (empty): however, very low expression for p35S-cel strain could be detected by using 0.5 µL cDNA. Higher cDNA concentration for T5-cel in RT-PCR displayed a higher expression, compared with the previous RT-PCR test, where lower cDNA concentration was used. Similar RT-PCR was performed with alcR primers to check for alcR gene expression in palcA-cel-alcR and palcA-gfp-alcR strains. We observed a strong amplification of p35S-alcR construct in both E. coli strains (A) and (B)).
Figure 3. Analysis of RT-PCR with gel electrophoresis. Transcription of the three genes (gfp, cellulase and alcR) targeted by correlating detection primers. cDNA (0.3 µL) was used for this test. (A) Transcription of cellulase gene for various strains (1% and 2% indicate ethanol concentration added to the culture). cDNA (0.5 µL) was used for this test (B).
To accurately determine the transcriptional signals efficiency, the cellulase gene expression was studied in E. coli by using qPCR. At present, qPCR is the most common and reliable method for quantitative analysis of nucleic acids.[Citation24] It has become one of the most reliable technologies for the analysis of gene expression.[Citation25] A method for comparative gene expression analysis is the evaluation of a target gene expression relative to an endogenous control gene (quantification of normalized expression values ΔΔCt).[Citation26] The expression of cellulase gene in E. coli was determined by using cysG gene (encoding uroporphyrin-III C-methyltransferase) as endogenous control, selected for normalization.
The studied constructs that are outlined in consist of: three derivatives of plasmid pBIN carrying alcA and CaMV 35S promoters (palcA-cel, p35S-cel and p35S-gfp), and pQE carrying coliphages T5 promoter (pT5 and pT5-cel). The three examined promoters can be arranged in a hierarchy, based on their activities in vivo under defined conditions (). The 35S promoter was located at the lower end of the hierarchy, indicating the lowest expression level among all the promoters in E. coli. Previous studies have reported that CaMV 35S promoter (the most common promoter used for generation of transgenic plants) can be recognized in E. coli with low expression efficiency,[Citation27,Citation28] although, theoretically, plant-specific regulatory systems are not supposed to function in bacteria due to difference in transcription regulatory system. Despite our RT-PCR results that showed very low cellulase expression for p35S-cel strain, no detectable transcription was found for p35S-cel in our qPCR experiment.
Figure 4. Expression comparison of cellulase genes between different E. coli strains. Clustergram showing all sorted samples, based on the expression intensity (red demonstrate the highest expression level and green the lowest level) (A). Expression variations between different E. coli strains (B).
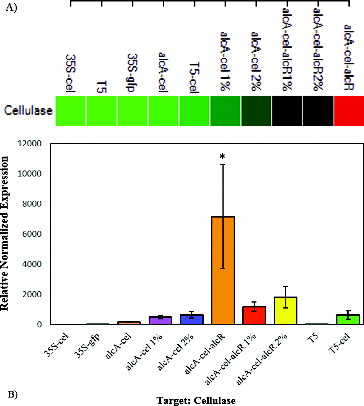
The strongest promoter identified in this study was alcA in palcA-cel-alcR strain, which was 11 times stronger than the T5 promoter (). The presence of AlcR transcription factor seemed to be critical for the functionality of the alcA promoter. This was mainly deduced from the data, where the cellulase gene expression level in palcA-cel-alcR strain was 37 times higher than palcA-cel line. Although alcR gene was regulated by 35S promoter, utilization of pCAMBIA (a high copy number plasmid) seemed to improve the transcription efficiency. This was confirmed by preliminary RT-PCR test indicating the absence of expression for p35S-cel cassette in pBIN plasmid, while exhibiting alcR gene expression in p35S-alcR cassette in pCAMBIA. The reason for this difference might be due to the plasmid copy number. pBIN is a low-copy-number plasmid and pCAMBIA is a high-copy-number plasmid, which may lead to higher expression of alcR gene in the latter plasmid.
We observed low non-induced basal level of alcA expression in the absence of AlcR. On the other hand, data show high ethanol dose-dependent expression of alcA promoter in the absence of AlcR activator. It has been reported that ethanol induction of the alcA promoter is dose-dependent.[Citation23,Citation29] Our transcription data also confirmed dose-dependent expression of alcA promoter by using ethanol. Cellulase gene expression in palcA-cel 2% and palcA-cel-alcR 2% was 1.3 and 1.5 times higher than palcA-cel 1% and palcA-cel-alcR 1% strains, respectively, although this data was not statistically significant. On the other hand, palcA-cel-alcR 1% and palcA-cel-alcR 2% strains exhibited 2.4 and 2.8 times higher expression levels than palcA-cel 1% and palcA-cel 2%, respectively. Our results reflected that the alcA promoter in E. coli is able to function in the absence of AlcR activator with lower efficiency; however, it displayed a non-significant dose-dependent activity in the presence of ethanol.
Analysis of alcR gene expression in palcA-cel-alcR strain
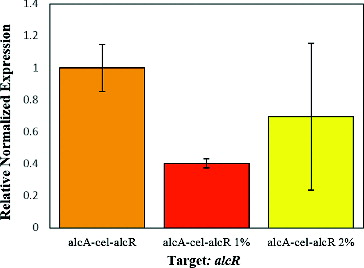
Transcription of alcR gene was also studied by qPCR to evaluate the expression level of alcR under the control of 35S promoter and to investigate the correlations between alcR expression and alcA activity. The results showed that the alcR gene expression trend in palcA-cel-alcR strain in the absence/presence of ethanol was very similar to the expression pattern of cellulase gene in this strain (). This data may display the direct correlation between AlcR concentrations and alcA activity, indicating the key role of the activator AlcR in the regulation of alcA promoter. On the other side, presence of AlcR increased the transcription level 37 times in the absence of ethanol, although this effect was significantly reduced when ethanol was added. Our data, which indicated a high non-induced basal level of alcA in the presence of AlcR, was consistent with the reported data from the study of heterologous alcR-alcA system in transformed Aspergilus niger.[Citation16] Their study showed clear differences in non-induced expression levels in both transcription and translation between A. nidulans and transformed A. niger. As they have suggested, this effect may be due to the accumulation of acetaldehyde from the regular metabolism.[Citation16] The same explanation may be applied to our results in E. coli, wherein acetaldehyde can be produced through many other cellular pathways, including ethanolamine utilization, preQ0 biosynthesis, threonine degradation IV, etc.[Citation30] Furthermore, high inducibility of alc system in A. nidulans may arise from the fine-tuning of alcA and aldA expression through maintaining an optimal acetaldehyde levels between inducing and toxic concentrations.[Citation31] Constitutive overexpression of the alcR gene, occurring in the gpdA : alcR transformants, for example,[Citation16] may disturb this balance and result in high non-induced basal expression. The activity of alcA promoter seemed to be directly related to the AlcR concentrations ( and ). Reduction in ethanol-induced gene expression in alcR-alcA strains may be explained by deleterious effects of ethanol, mainly on alcR expression. These changes on transcription and translation may be due to the direct effect of ethanol on the conformation of RNA polymerase and ribosomes.[Citation32] Another explanation of these results might be the ethanol-inducing effect of heat shock response in bacteria by the induction of RNA polymerase sigma factor σ32.[Citation33] Lower expression of alcR-aclA system in the presence of ethanol might be associated with the competition between different transcription sigma factors, especifically σ70 and σ32. Sigma factor 32 might have greater affinity for RNA polymerase core enzyme than sigma factor 70 during the heat shock response.[Citation34] Increase in the transcription level of ethanol induced alcA-cel-alcR strain may also be related to the increase in the activity of σ32. In addition to σ32, other heat shock related sigma factors may also be involved in ethanol induced alcA transcription. Further experiments are required to support these hypotheses and clarify the mechanism of the alcR-alcA system in E. coli.
Figure 5. Expression comparison of alcR gene under different ethanol concentrations.
Transient expression in tobacco
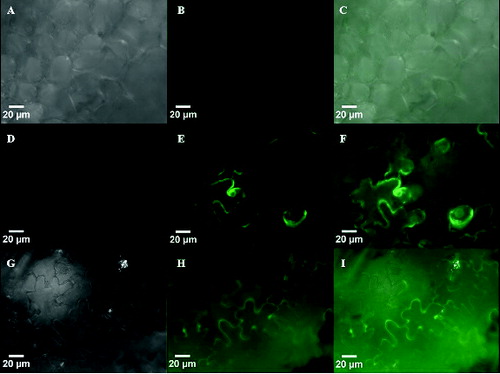
Application of alcA promoter has already been well established in plants.[Citation14,Citation15,Citation35] The alcR-alcA system has been modified to increase the efficiency of this system in transgenic plants. This modification comprises the regulation of alcR gene under the control of CaMV 35S promoter and construction of alcAmin 35S promoter, in which two AlcR binding sites are attached to the CaMV 35S minimal promoter.[Citation15] It has been reported that this inducible system is tightly regulated and shows negligible basal activity in plants.[Citation14,Citation15] In this study, we showed that alcA promoter has high ethanol-induced expression level in the absence of AlcR. To detect the possibility of such occurrence in plants, we performed transient expression assay by using a native alcA promoter from A. nidulans in tobacco. We also characterized the time course of induction and gene expression of alcA compared with 35S promoter. Tobacco leaves were infiltrated with Agrobacterium carrying both palcA-gfp and p35S-alcR constructs as well as Agrobacteria harbouring individual cassette of p35S-gfp, palcA-gfp and p35S-alcR. It has been reported that the best concentration of ethanol for induction of the promoter is 2% ethanol (v/v),[Citation14,Citation36] which was used in this study as well. The fluorescent activity was observed for leaves treated with CaMV 35S promoter after three days and for alcA promoter induced with ethanol after five days from the time of infiltration (). The gfp expression lasted for more than 10 days. Like in the wild type, no fluorescent activity was observed in treated leaves with either palcA-gfp or p35S-alcR, irrespective of the presence of ethanol. No expression was also observed in alcA-gfp-alcR with no ethanol induction (data not shown). These data suggested that the native alcA regulon was fully functional and inducible by ethanol in tobacco. Results showed no fluorescent activity for alcA promoter when alcR gene was not present. This may indicate no or negligible basal activity for this promoter. Our results revealed faster expression of the constitutive promoter over the inducible promoter. It has been previously shown that the ethanol induction of alcA promoter in transgenic plant is fast.[Citation14,Citation15] In our experiments, the delayed expression of alcA promoter may be due to the time requirement for the production of AlcR protein and this delay may not be due to lack of minimal 35S sequence in alcA. Further research is needed to confirm the reason for the delay in the alcA promoter activity.
Figure 6. Epifluorescence micrographs of GFP fluorescence in tobacco leaves expressing p35S-gfp at day three (D–F) and palcA-gfp-alcR at day five (G–I). Fluorescent activity of these lines was compared to wild type leaves (A–C), which showed no fluorescence.
Conclusions
The ethanol utilization pathway in A. nidulans is a highly inducible regulatory system for consumption of ethanol as a carbon source. The pathway requires a specific activator (AlcR encoded by alcR gene) mediating the transcription in the presence of an inducing compound (e.g. ethanol). We showed that the native alcA regulon from A. nidulans is fully functional and tightly regulated in tobacco. This alcA induction was only obtained in the presence of both AlcR and ethanol. Our results show the higher efficiency of alcA promoter over T5 and 35S promoter in E. coli. The palcA-cel-alcR strain exhibited the highest transcription level among all strains, irrespective of ethanol induction. Except palcA-cel and palcA-cel 1%, all other strains carrying alcA promoter showed higher transcription levels compared with the T5-cel promoter. This study suggests that high levels of expression of heterologous genes can be obtained in E. coli using alcR-alcA system of A. nidulans. It can also be concluded that slightly different mechanisms may be involved in the regulation of alc system in A. nidulans and plants, compared with E. coli. We recommend the broader application of alcR-alcA system in E.coli using high copy number plasmids for mass production of cellulase enzyme and bioethanol.
Supplemental data
Supplemental data for this article can be accessed at http://dx.doi.org/10.1080/13102818.2015.1065711.
Supplementary_file.pdf
Download PDF (299.6 KB)Acknowledgements
We would like to sincerely thank Syngenta, USA for providing us the plasmids carrying alcA promoter and alcR gene. We would like to specially thank Dr. Ernest Kwok, California State University, Northridge for providing us the tobacco plant and for his assistance in microscopy.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Alvira P, Tomás-Pejó E, Ballesteros M, et al. Pretreatment technologies for an efficient bioethanol production process based on enzymatic hydrolysis: a review. Bioresour Technol. 2010;101(13):4851–4861.
- Sun J, Zhou X. Utilization of lignocellulose-feeding insects for viable biofuels: an emerging and promising area of entomological science. In: Liu T, Kang L, editors. Recent advances in entomological research. From molecular biology to pest management. Heidelberg: Springer; 2011. p. 434–500.
- Edwards MC, Henriksen ED, Yomano LP, et al. Addition of genes for cellobiase and pectinolytic activity in Escherichia coli for fuel ethanol production from pectin-rich lignocellulosic biomass. Appl Environ Microbiol. 2011;77 (15):5184–5191.
- Bokinsky G, Peralta-Yahya PP, George A, et al. Synthesis of three advanced biofuels from ionic liquid-pretreated switchgrass using engineered Escherichia coli. Proc Natl Acad Sci USA. 2011;108(50):19949–19954.
- Steen EJ, Kang Y, Bokinsky G, et al. Microbial production of fatty-acid-derived fuels and chemicals from plant biomass. Nature. 2010;463(7280):559–562.
- Ibrahim E, Jones KD, Hossenya EN. Molecular cloning and expression of cellulase and polygalacturonase genes in E. coli as a promising application for biofuel production. J Pet Environ Biotechnol. 2013;4(3):1000147.
- Atsumi S, Cann AF, Connor MR, et al. Metabolic engineering of Escherichia coli for 1-butanol production. Metab Eng. 2008;10(6):305–311.
- Hanai T, Atsumi S, Liao JC. Engineered synthetic pathway for isopropanol production in Escherichia coli. Appl Environ Microbiol. 2007;73(24):7814–7818.
- Ingram LO, Aldrich HC, Borges AC, et al. Enteric bacterial catalysts for fuel ethanol production. Biotechnol Prog. 1999;15(5):855–866.
- Dien BS, Cotta MA, Jeffries TW. Bacteria engineered for fuel ethanol production: current status. Appl Microbiol Biotechnol. 2003;63(3):258–266.
- Flipphi M, Kocialkowska J, Felenbok B. Characteristics of physiological inducers of the ethanol utilization (alc) pathway in Aspergillus nidulans. Biochem J. 2002;364(Pt 1):25–31.
- Mathieu M, Fillinger S, Felenbok B. In vivo studies of upstream regulatory cis-acting elements of the alcR gene encoding the transactivator of the ethanol regulon in Aspergillus nidulans. Mol Microbiol. 2000;36(1):123–131.
- Battaglia R, Brambilla V, Colombo L, et al. Functional analysis of MADS-box genes controlling ovule development in Arabidopsis using the ethanol-inducible alc gene-expression system. Mech Dev. 2006;123(4):267–276.
- Roslan HA, Salter MG, Wood CD, et al. Characterization of the ethanol-inducible alc gene-expression system in Arabidopsis thaliana. Plant J. 2001;28(2):225–235.
- Caddick MX, Greenland AJ, Jepson I, et al. An ethanol inducible gene switch for plants used to manipulate carbon metabolism. Nat Biotechnol. 1998;16(2):177–180.
- Nikolaev I, Mathieu M, van de Vondervoort P, et al. Heterologous expression of the Aspergillus nidulans alcR-alcA system in Aspergillus niger. Fungal Genet Biol. 2002;37 (1):89–97.
- Panozzo C, Capuano V, Fillinger S, et al. The zinc binuclear cluster activator AlcR is able to bind to single sites but requires multiple repeated sites for synergistic activation of the alcA gene in Aspergillus nidulans. J Biol Chem. 1997;272(36):22859–22865.
- Pang H, Zhang P, Duan CJ, et al. Identification of cellulase genes from the metagenomes of compost soils and functional characterization of one novel endoglucanase. Curr Microbiol. 2009;58(4):404–408.
- Haseloff J, Siemering KR, Prasher DC, et al. Removal of a cryptic intron and subcellular localization of green fluorescent protein are required to mark transgenic Arabidopsis plants brightly. Proc Natl Acad Sci USA. 1997;94(6):2122–2127.
- Mattanovich D, Rüker F, Machado AC, et al. Efficient transformation of Agrobacterium spp. by electroporation. Nucleic Acids Res. 1989;17(16):6747.
- Murashige T, Skoog F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant. 1962;15(3):473–497.
- Yang Y, Li R, Qi M. In vivo analysis of plant promoters and transcription factors by agroinfiltration of tobacco leaves. Plant J. 2000;22(6):543–551.
- Lee S, Oh E, Oh Y, et al. Increased ethanol resistance in ethanolic Escherichia coli by Insertion of heat-shock genes BEM1 and SOD2 from Saccharomyces cerevisiae. Biotechnol Bioprocess Eng. 2010;15(5):770–776.
- Sochivko DG, Fedorov AA, Varlamov DA, et al. Simulation of the PCR amplification as two-type-particle branching process. Dokl Biochem Biophys. 2010;434:239–241.1
- Ginzinger DG. Gene quantification using real-time quantitative PCR: an emerging technology hits the mainstream. Exp Hematol. 2002;30(6):503–512.
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29 (9):e45.
- Assaad FF, Signer ER. Cauliflower mosaic virus P35S promoter activity in Escherichia coli. Mol Gen Genet. 1990;223(3):517–520.
- Lewin A, Jacob D, Freytag B, et al. Gene expression in bacteria directed by plant-specific regulatory sequences. Transgenic Res. 1998;7(6):403–411.
- Chong H, Huang L, Yeow J, et al. Improving ethanol tolerance of Escherichia coli by rewiring its global regulator cAMP receptor protein (CRP). PLoS One. 2013;8(2):e57628.
- Keseler IM, Mackie A, Peralta-Gil M, et al. EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res. 2013;41(Database issue):D605–D612.
- Felenbok B, Flipphi M, Nikolaev I. Ethanol catabolism in Aspergillus nidulans: a model system for studying gene regulation. Prog Nucl Acid Res Mol Biol. 2001;69:149–204.
- Haft RJ, Keating DH, Schwaegler T, et al. Correcting direct effects of ethanol on translation and transcription machinery confers ethanol tolerance in bacteria. Proc Natl Acad Sci U S A. 2014;111(25):E2576–E2585.
- Van Dyk TK, Reed TR, Vollmer AC, et al. Synergistic induction of the heat shock response in Escherichia coli by simultaneous treatment with chemical inducers. J Bacteriol. 1995;177(20):6001–6004.
- Blaszczak A, Zylicz M, Georgopoulos C, et al. Both ambient temperature and the DnaK chaperone machine modulate the heat shock response in Escherichia coli by regulating the switch between sigma 70 and sigma 32 factors assembled with RNA polymerase. EMBO J. 1995;14 (20):5085–5093.
- Deveaux Y, Peaucelle A, Roberts GR, et al. The ethanol switch: a tool for tissue-specific gene induction during plant development. Plant J. 2003;36(6):918–930.
- Klose H, Günl M, Usadel B, et al. Ethanol inducible expression of a mesophilic cellulase avoids adverse effects on plant development. Biotechnol Biofuels. 2013;6(1):53.