
ABSTRACT
Integrons play an important role in multidrug resistance. The integron platform codes for integrase (intI) that is required for gene cassette integration through site-specific recombination. The recombination crossover occurs between the G and TT nucleotides in non-palindromic attI and palindromic attC sites. The aim of this study was to establish an efficient in vitro assay for integrase purification and activity detection. To this end, the intI gene was cloned into the pET-22b plasmid. Then, the resulting recombinant plasmid was transformed into Escherichia coli Origami™ strain. The recombinant protein expression was confirmed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blot assays. The recombinant intI protein was purified by nickel–nitrilotriacetic acid (Ni–NTA) affinity chromatography, and its activity was measured by a newly introduced assay. Briefly, specific primers for each side of attI and attC were used, thereby, a polymerase chain reaction would be performed, if a fused plasmid containing both attI and attC sites was created upon recombination. SDS-PAGE and western blotting confirmed the presence of a 38-kDa recombinant protein. Optimum conditions were established for the measurement of the integrase activity and a new model assay was conducted to analyse the recombination activity in vitro. Although the electrophoretic mobility shift assay is an efficient and reliable method, the newly introduced assay provided new or enhanced capability to determine the integrase activity, suggesting that there is no need for expensive and advanced equipment.
Introduction
Overuse of antimicrobial agents is considered to be responsible for the emergence of multidrug resistance. Integrons are functional DNA and bacterial genetic elements, which play an important role in multidrug resistance among Gram-negative and some Gram-positive bacteria.[Citation1–3] There are at least three classes of integrons, in which class 1 integrons represent the most widespread class of mobile DNA elements in the resistant bacterial strains. All class 1 integrons are composed of three essential parts: (1) an integrase gene (intI) encoding an integrase, which is a member of the tyrosine recombinase family with a site-specific recombination activity, (2) non-palindromic attI and palindromic attC recombination sites specific to each integrase and (3) one or more mobile gene cassettes.[Citation1] DNA elements, such as integrons, have the ability to acquire and transfer gene cassettes (most antibiotic resistance genes) between Gram-negative bacteria. Cassettes are preferentially transferred by a mechanism known as conservative site-specific recombination at the attI site, containing the core site GTTRRRY.[Citation4]
The highly conserved intI gene, encoding an integrase, is demonstrated to play a main role in capturing and spreading of antibiotic resistance genes by integrons. Integrase, a 337-amino acid enzyme, is required for gene cassette integration through site-specific recombination.[Citation5]
There are two conserved sequences and one variable region among class 1 integrons. The 5' conserved sequences (CS) of class 1 integrons contain the intI gene, an attachment site for recombination (attI) and a promoter,[Citation6] whereas, 3' conserved sequences (3' CS) possess other genes covering an antiseptic resistance gene (qacEΔ1), a sulphonamide resistance gene (sulI) and an open reading frame (ORF) of unknown function.[Citation7] A variable genomic region is found between two conserved sequences, containing 0–8 specific cassettes.[Citation5,Citation8] Although carrying defined antibiotic resistance genes, the cassettes can be captured through recombination and re-inserted in the gene structure with a new arrangement of the cassettes, a process called reintegration.[Citation7,Citation9] Therefore, a number of deaths are attributed to broad antibiotic resistance to different drugs.[Citation10] In general terms, cassettes consist of attC sites, so-called 59-base elements, at the 3' end of the ORF.[Citation11] The 59-base element sites share regions of about 25 bp at each end, in spite of significantly divergent nucleotide sequences and lengths. The attC sites, which are required for in vivo recombination activity,[Citation12–14] are composed of two imperfect inverted repeats with a 7 bp core site GTTRRRY.[Citation13,Citation15,Citation16] The recombination crossover occurs between the G and T nucleotides in attC and attI sites.[Citation14,Citation17]
Several in vitro biochemical assays have been developed to measure the enzymatic activity of bacterial and viral integrases either alone or in combination with various inhibitors.[Citation18–21] Traditionally, bacterial and viral integrase activity has been measured by low-throughput gel-based method involving radioactively labelled oligonucleotides known as the electrophoretic mobility shift assay (EMSA).[Citation22–24]
However, there is always a need for rapid diagnostic tools to exert highly reliable modern technologies for an accurate measurement of integrase activity among multidrug-resistant bacterial strains. In light of this, the purpose of the present study was to develop a simple in vitro assay, allowing the detection of recombinant products derived from the integrase recombination activity on attI and attC sequences. However, it is clear that further studies are needed to find integrase inhibitors and other biochemical properties.
Materials and methods
Bacterial strains and intI gene amplification
The integrase positive strain Klebsiella pneumoniae PTCC 1290 was obtained from Persian Type Culture Collection. The integrase sequence was retrieved from the National Center for Biotechnology Information (NCBI) with accession number LN864819.1, to design specific polymerase chain reaction (PCR) primers.
Genomic DNA was extracted by using a high pure PCR Template Preparation Kit (Roche, Germany Construction Co). PCR (Master Cycler, Eppendorf, Germany) was performed to amplify a 1,014 bp fragment of the highly conserved integrase region. To facilitate cloning, NdeI and XhoI restriction enzyme sites were introduced into the 5' end of the forward and reverse primers, respectively. The PCR reaction was performed in a final volume of 25 μL containing 1 μL DNA, 0.4 mmol/L dNTP, 2 mmol/L MgCl2, 200 nmol/L of each intI specific forward and reverse primers (intI F: 5'-AACATATGAAAACCGCCACTGCGCCGTTACCAC-3' and intI R: 5'-AAACTCGAGCTCACTAGTGAGGGGCGGCAGCG-3') and 1.5 U Taq DNA Polymerase (Cinnagen, Iran). The PCR amplification was carried out under the following conditions: an initial denaturation at 94 °C for 5 min, 32 cycles of denaturation at 94 °C for 1 min; annealing at 65 °C for 1 min, elongation at 72 °C for 1.5 min and a final extension at 72 °C for 10 min.
Construction of an expression vector containing the intI gene
The PCR product was checked on 1.5% agarose gel by running an electrophoresis (Bio-Rad) in 1× Tris-acetate-ethylenediaminetetraacetic acid buffer for 1 h at 80 V and visualized by using ethidium bromide. A 1 kb DNA ladder (Fermentase) was used to confirm the size of the product. To insert the intI fragment into the plasmid, the resultant 1,014 bp PCR product was purified by using a Gel Extraction Kit (Fermentas Inc.) according to the manufacturer's instructions. The recovered PCR product and pET-22b plasmid were digested with 20 U of NdeI and XhoI restriction enzymes (Fermentase) and ligated with 20 U of T4 Ligase (Fermentase) overnight at 16 °C. The ligation mixture was transformed into Escherichia coli Top10F' competent cells by using the CaCl2 heat shock method at 42 °C for 60 s. The presence of the correctly sized insert was verified by colony PCR using T7 promoter and T7 terminator primers. The PCR mix contained 0.5 μg of DNA, 0.1 mmol/L dNTP, 50 mmol/L KCl, 10 mmol/L Tris (pH 8.3), 1.5 mmol/L MgCl2, 200 nmol/L of each of the specific forward (T7 promoter: 5'-TAATACGACTCACTATAGGG-3') and reverse (T7 terminator 5'-GCTAGTTATTGCTCAGCGG-3') primers and 1.25 U of Taq DNA Polymerase (Cinnagen, Iran) in a final volume of 50 μL. PCR amplification initially started with a 95 °C initial denaturation for 3 min, followed by 25 cycles of 30 s at 95 °C (denaturation), 30 s at 65 °C (annealing) and 30 s at 72 °C (extension) with a final extension at 72 °C for 5 min. To confirm the digestion, a single colony was cultured in Luria–Bertani (LB) agar (Merck, Germany) and the bacterial plasmid was extracted using the Plasmid Extraction Kit (Vivantis). The transformants harbouring the pET22-intI construct were digested with NdeI and XhoI restriction enzymes. Briefly, pET22-intI recombinant expression vector was digested with 10 U of each of XhoI and NdeI (Fermentase) at 37 °C for 3 h. The digested products were analysed again on 1% agarose gel and visualized by using ethidium bromide. Authenticity of the final construct was confirmed by DNA sequencing (Genfanavaran, Macrogen, Seoul, Korea) of extracted plasmids with T7 promoter and T7 terminator universal primers.
Recombinant expression of intI
The colony containing pET22-intI plasmid was extracted from E. coli Top10F' by Plasmid Extraction Kit (Vivantis) and transformed into E. coli Origami™ strain. A fresh bacterial colony harbouring pET22-intI was inoculated into LB broth medium (Merck, Germany) and incubated at 37 °C in a shaker incubator until the bacterial growth reached log phase (optical density (OD) at 600 nm = 0.5–0.9 using BioTek Epoch spectrophotometer). The protein expression was induced in log phase with different concentrations (0.5–1 mmol/L) of isopropyl β-D-1-thiogalactopyranoside (IPTG). Samples were collected before and after induction and were allowed to grow for 12–14 h at 37 °C in a shaker (Heidolph) at 180 revolutions per minute (rpm). The desired enzyme was expressed in fusion with 6×His-tag at the 3' end in E. coli Origami strain. Protein expression was analysed with 15% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blot.[Citation25] SDS-PAGE was performed by using 15% polyacrylamide gel in a vertical slab gel apparatus (Bio-Rad Instrument). The purified integrase mixed with the same volume of loading buffer was boiled at 100 °C for 10 min and subsequently subjected to SDS-PAGE at 100 V for 15 min and then at 200 V for 45 min. The gel was stained with Coomassie Brilliant Blue G-250 and destained with the destaining solution (2.5% methanol and 10% acetic acid) for 3–5 h. PageRuler™ Prestained Protein Ladder (Thermo Scientific, MA, USA) was used in these experiments.
Purification of the recombinant intI protein
Freshly cultured transformed E. coli Origami™ was inoculated in 500 mL LB broth (Merck, Germany) containing ampicillin (50 μg/mL) and incubated at 37 °C for 12 h in a shaker (Heidolph) at 180 rpm. The bacterial growth was assessed by turbidity measurement at 600 nm OD. When the OD at 600 nm reached 0.4, IPTG, as a protein expression inducer, was added to the culture medium and incubated at 25 °C overnight. Bacterial pellets were collected by centrifugation at 4,000 rpm for 15 min (ALC 4239R centrifuge, ALC, Milan, Italy). The pellets were resuspended in 5 mL lysis buffer I (50 mmol/L NaH2PO4, pH 7.5, 1% Sigma protease inhibitor cocktail P-8340 and 50 mg/mL lysozyme), incubated at room temperature for 20 min and further centrifuged at 4,000 rpm for 15 min. Subsequently, the pellets were resuspended in 5 mL lysis buffer II (50 mmol/L NaH2PO4, pH 7.5, 500 mmol/L NaCl, 1 mmol/L dithiothreitol (DTT) and 0.025% Triton X-100). Then, the bacterial suspension was sonicated (Dr Hielscher, GmbH, Germany; UP 200S) 10 times, each for 25 s. The sample was centrifuged at 10,000 rpm for 25 min to pellet the cell debris; the supernatant was collected, filtered through a 0.45-micron membrane filter (Biofil) and used directly as a template for further purification, followed by SDS-PAGE.
The clarified supernatant was directly purified using a nickel–nitrilotriacetic acid (Ni–NTA) affinity column according to the instructions supplied by the manufacturer (QIAGEN, Germany). Briefly, after washing the column with wash buffer I (50 mmol/L NaH2PO4, pH 7.5), the supernatant was loaded onto the column and the column was washed with wash buffer II (50 mmol/L NaH2PO4, pH 7.5, 500 mmol/L NaCl, 0.025% Triton X-100 and 20 mmol/L imidazole). The integrase protein was eluted from the column by using elution buffer (50 mmol/L NaH2PO4, pH 7.5, 500 mmol/L NaCl, 0.025 Triton X-100 and 400 mmol/L imidazole). The eluted fraction was dialysed against phosphate-buffered saline (PBS) 50 mmol/L, pH 7.5, and the purity of the integrase protein was assessed by 15% SDS-PAGE and western blot. SDS-PAGE was performed as described earlier and western blot was performed using (anti-His) specific anti-intI monoclonal antibody (Sigma-Aldrich). The protein concentration was measured by the Bradford assay and the purified intI protein was frozen with 10% glycerol and stored at −20 °C until further use.
Integrase activity measurement
To examine the integrase functional activity, 1 μg of the purified intI enzyme was incubated with the substrate (250 ng of both attI and attC fragments in 20 mmol/L Tris–HCl buffer (pH = 7.5) containing 100 mmol/L NaCl, 0.1 mmol/L ethylenediaminetetraacetic acid and 1% glycerol). The nucleotide sequences of attI and attC were as follows: 5'-ACGCCGTGGGTCGATGTTTGATGTTATGGAGCAGCAACGATGTTACGCAGCAGGGCAGTCGCCCTAAAACAAAGTTAGGTGGCTCAATGAGCATCATTGC-3' and 5'-CGCCCGTCTAACAATTCGTTCAAGCCGACGTTGCTTCGTGGCGGCGCTTGCGTGCTACGCTAAGCTTCGCACGCCGCTTGCCACTGCGCACCGCGGCTTAACTCAGGCGTTAGATGCACT-3', respectively. The sequences were introduced into pBSK (+) Simple-Amp and pBSK(+) Simple-Kan vectors. The reaction mixture with a final volume of 20 μL was incubated first at 4 °C for 20 min and then at 37 °C for 1 h, and finally, heat treatment was performed at 80 °C for 20 min to inactivate the mixture.
PCR-based detection of in vitro site-specific recombination
Two specific primers (P1 and P2) were designed for the amplification of attI and attC genes to detect the integrase activity. The PCR (Master Cycler, Eppendorf, Germany) was conducted under the following conditions: initial denaturation at 94 °C for 5 min and 35 cycles of denaturation at 94 °C for 30 s; annealing at 52 °C for 30 s; elongation at 72 °C for 1 min and a final extension at 72 °C for 5 min. The specific primers P1 and P2 used to check the in vitro recombination reaction were as follows: P1 (F): 5'-CGCCCGTCTAACAATTCG-3' and P2 (R): 5'-CGAGGCAATGATGCTCATTG-3'. The PCR reaction was carried out in a final volume of 25 µL containing 10 pmol of each primer, 10 mmol/L Tris, pH 8.3, 0.2 mmol/L dNTP, 1.5 mmol/L MgCl2, 2.5 U Taq Polymerase and 1 µL integrase reaction mixture, as a DNA template. The post-recombination PCR product was checked on 1.5% agarose gel by running electrophoresis (Bio-Rad) in 1× Tris-acetate-ethylenediaminetetraacetic acid buffer for 1 h at 80 V and visualized by using ethidium bromide. A 1 kb DNA ladder (Fermentase) was used to confirm the size of the product.
Results and discussion
Cloning of the intI coding sequence
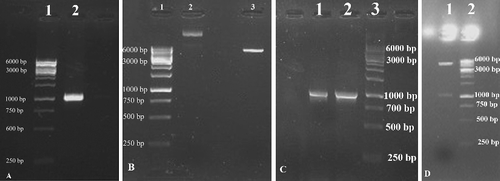
The integrase gene was amplified with specific primers under the conditions described earlier. As shown in (A), the PCR yielded an amplified product with an expected size of 1,014 bp, checked by using 1.5% gel electrophoresis. The amplified PCR product was digested with NdeI and XhoI restriction enzymes and subsequently ligated into the expression vector pET-22b digested with the same restriction enzymes to create the pET22-intI recombinant vector ((B)). Recombinant clones were confirmed by a colony PCR using T7 promoter and T7 terminator primers, as depicted in (C). The recombinant plasmid pET22-intI was confirmed by double digestion with NdeI and XhoI restriction enzymes and DNA sequencing, as shown in (D).
Figure 1. Agarose gel electrophoresis of the PCR amplified integrase gene (A); pattern of restriction enzyme digestion of pET-22b containing intI fragment (B); colony PCR of top10F' transformants (C); digested recombinant plasmid pET22-intI (D).
Protein expression and purification analysis
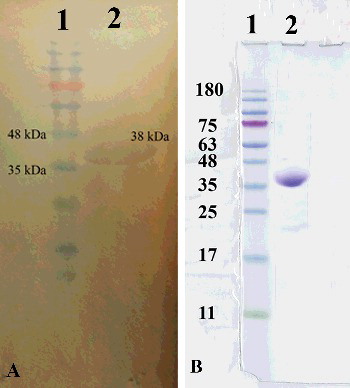
Protein expression was induced by the addition of IPTG. As illustrated in (A), expression of the intI enzyme was clearly identified by a 38-kDa band on an SDS-PAGE gel and validated by western blot. Because of the presence of 6xHis-tag in the C-terminus of the protein, Ni–NTA affinity chromatography was used to purify the intI protein ((B)). The integrase concentration after the purification step was determined by the Bradford assay and was found to be 0.5 mg/mL.
Figure 2. Integrase expression with western blot analysis using (anti-His) specific anti-intI monoclonal antibody (A) and purification of C-terminal 6xHis-tagged intI under native conditions with Ni–NTA method (B).
Integrase activity measurement
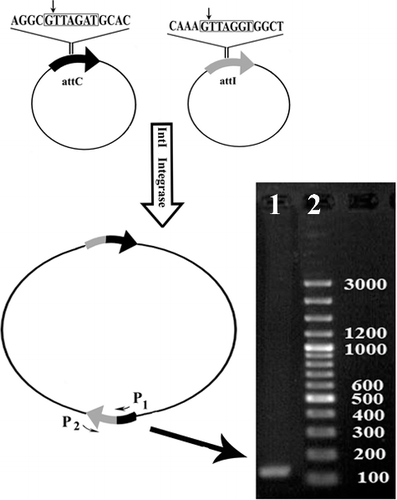
The integrase activity was evaluated by excision and integration between two plasmids, including pBSK (+) Simple-Amp and pBSK (+) Simple-Kan, carrying the full-length sequences of attI and attC in the suitable direction. Recombination between the two plasmids yielded a recombinant fragment. Following the evaluation of intI recombination activity, a PCR screening test was performed, yielding a 147 bp fragment (). The findings from this study demonstrated that the resultant integrase was active for site-specific integration between attI and attC.
Figure 3. Schematic representation of recombination pattern of two vectors containing attI and attC sequences recognized by intI by using specific primers.
Integrase enzyme and recombination pattern
The spread of antibiotic resistance among Gram-negative bacteria is mediated by mobile elements such as integrons. In general, integrons contain integrase (intI) that has the ability to perform site-specific recombination by capturing and excising gene cassettes.[Citation18,Citation26] Gene cassette mobility between various integrons, which is mediated by integrase, is a leading cause of multidrug resistance in clinical settings, resulting in limited success in the treatment of patients receiving long-term antibiotic therapy.[Citation27] Due to the key role of integrase in the spread of antibiotic resistance genes among bacterial strains and the rapid appearance of multidrug resistance, we attempted to purify the integrase protein from K. pneumoniae. In the present study, protein expression was induced by different concentrations of IPTG. Our results indicated that the expression level reached its highest rate at the maximal concentration of 1 mmol/L IPTG. However, other IPTG concentrations were not enough to have good over-expression of the recombinant integrase. It seemed that the promoter saturation by the optimal IPTG concentration is a possible reason why higher concentrations of IPTG failed to induce the expression. In order to prevent excessive accumulation of the recombinant protein and the formation of insoluble aggregates, intI expression was induced by the addition of IPTG at 25 °C.[Citation28] Following this treatment, the rate of bacterial protein expression began to decrease, but the solubility was improved. On one hand, integrase is a highly insoluble protein, and on the other hand, salt addition and reduction of temperature to 25 °C were demonstrated to improve the protein solubility.[Citation28,Citation29] For this reason, a higher dose of NaCl (500 mmol/L) was used to mimic the physiological conditions required for enzyme solubility. In the present study, it was shown that the E. coli Origami™ strain is a suitable host for the large-scale production of the recombinant intI protein. The successful expression of the recombinant protein was verified by the presence of the 1,014 bp fragment, found by using SDS-PAGE gel analysis. The enzyme activity was examined after purification. It is important to note that several factors should be considered when measuring an enzyme activity in order to have the active enzyme as close as possible to the native conditions. Enzyme and substrate concentrations have been shown to have a significant impact on the enzyme's ability to catalyse a specific reaction.[Citation30] Some detergents, including 0.025% Triton X-100 and DTT, are commonly used to improve the enzyme activity.
In the present study, the plasmid substrates were designed to generate a larger fused plasmid containing both attI and attC sites upon recombination. PCR amplification on the recombination product exhibited amplified DNA fragments with a typical length of 147 bp. This explains that recombination occurred when the integrase protein was added to the substrates. In a previous study, recombination analysis of intI between the attI and attC sites was performed by the EMSA, based on radiolabelled fragments.[Citation18] However, the new approach introduced in this study makes it possible to determine the integrase function by using unlabelled fragments present in subsequent PCR screening.[Citation19,Citation22,Citation23] Radioactively labelled oligonucleotide probes, such as footprinting and EMSA, although still commonly used, are expensive, laborious and time-consuming methods, with increased risk of radiation exposure.[Citation23,Citation31,Citation32] One of the main drawbacks of autoradiography is its requirement for highly expensive conditions. The mobility shift assay is potentially hazardous for the human health because of the presence of 32P-labelled nucleic acids. Moreover, several fluorescence-based assays, such as fluorescence resonance energy transfer, have also been reported for the measurement of integrase activity. Fluorescence-based assays require a dually labelled oligonucleotide probe, multiple labelling processes and advanced detection devices. Overall, these methods are limited by the need for large amounts of oligonucleotide substrates, processing steps and high cost. Therefore, there is a pressing need for the development of non-radioactive assays to measure the integrase activity. In our non-radioactive novel method, two vectors containing attI and attC sites were provided for determining the integrase recombination activity, as an alternative to bio-hazardous radiolabelled substrates. Thus, integrative and excessive recombination activities were detected without producing hazardous waste. Therefore, the results from this study demonstrated that PCR-based recombination is a suitable and inexpensive method for measuring the integrase activity and further analysis. Also, the PCR-based recombination assays can greatly facilitate further biochemical and structural analyses of the integrase.
It appears that expanded knowledge about the function and catalytic mechanisms of the intI enzyme can provide new insights into determination of its biochemical properties. However, since the ability of intI for successful integration was clearly demonstrated in this study, further research studies are needed to design novel integrase inhibitors for prevention of intI recombination. The availability of specific inhibitors can provide significant opportunities to solve major problems associated with antibiotic resistance spread.
Conclusions
Because of the importance of integron to capture, integrate and express gene cassettes encoding antibiotic resistance enzymes in clinical settings, we sought to investigate high-level expression, purification and novel method for activity measurement of histidine-tagged recombinant integrase. In the present study, an oligonucleotide vector assay with unlabelled fragment, as a substrate, was introduced as a novel method for assessing the activity of the purified intI enzyme. However, this study provided a fast and convenient method for the detection of integrase recombination activity, as a suitable alternative to the traditionally used radiolabelled substrate, which is hazardous for the human health. The assay can also be adapted to design specific inhibitors, which may help to reduce the spread of antibiotic resistance.
Disclosure statement
The authors declare that they have no conflict of interest.
Additional information
Funding
References
- Ishikawa S. Simultaneous PCR detection of multiple classes of integron integrase genes for determining the presence of multidrug-resistant bacteria in environmental samples. Curr Microbiol. 2011;62(6):1677–1681.
- Moradian Kouchaksaraei F, Ferdosi Shahandashti E, Molana Z, et al. Molecular detection of integron genes and pattern of antibiotic resistance in pseudomonas aeruginosa strains isolated from intensive care unit, Shahid Beheshti hospital, north of Iran. Int J Mol Cell Med. 2012;1(4):209–217.
- Rajabnia R, Asgharpour F, Ferdosi Shahandashti E, et al. Class 1 integron in Pseudomonas aeruginosa isolates from different places and devices of ICU in Babol, Iran. Jundishapur J Microbiol. 2013;6(2):eE4850.
- Larouche A, Roy PH. Effect of attC structure on cassette excision by integron integrases. Mobile DNA. 2011;2(1):3.
- Levesque C, Piche L, Larose C, et al. PCR mapping of integrons reveals several novel combinations of resistance genes. Antimicrob Agents Chemother. 1995;39(1):185–191.
- Bennett PM. Integrons and gene cassettes: a genetic construction kit for bacteria. J Antimicrob Chemother. 1999;43(1):1–4.
- Hall RM, Collis CM. Mobile gene cassettes and integrons: capture and spread of genes by site-specific recombination. Mol Microbiol. 1995;15(4):593–600.
- Naas T, Mikami Y, Imai T, et al. Characterization of In53, a class 1 plasmid- and composite transposon-located integron of Escherichia coli which carries an unusual array of gene cassettes. J Bacteriol. 2001;183(1):235–249.
- Stokes HW, Hall RM. A novel family of potentially mobile DNA elements encoding site-specific gene-integration functions: integrons. Mol Microbiol. 1989;3(12):1669–1683.
- Villa L, Pezzella C, Tosini F, et al. Multiple-antibiotic resistance mediated by structurally related IncL/M plasmids carrying an extended-spectrum beta-lactamase gene and a class 1 integron. Antimicrob Agents Chemother. 2000;44(10):2911–2914.
- Recchia GD, Hall RM. Gene cassettes: a new class of mobile element. Microbiology (Reading, England). 1995;141(12):3015–3027.
- Collis CM, Hall RM. Gene cassettes from the insert region of integrons are excised as covalently closed circles. Mol Microbiol. 1992;6(19):2875–2885.
- Hall RM, Brookes DE, Stokes HW. Site-specific insertion of genes into integrons: role of the 59-base element and determination of the recombination cross-over point. Mol Microbiol. 1991;5(8):1941–1959.
- Stokes HW, O'Gorman DB, Recchia GD, et al. Structure and function of 59-base element recombination sites associated with mobile gene cassettes. Mol Microbiol. 1997;26(4):731–745.
- Collis CM, Hall RM. Expression of antibiotic resistance genes in the integrated cassettes of integrons. Antimicrob Agents Chemother. 1995;39(1):155–162.
- Recchia GD, Stokes HW, Hall RM. Characterisation of specific and secondary recombination sites recognised by the integron DNA integrase. Nucleic Acids Res. 1994;22(11):2071–2078.
- Hansson K, Skold O, Sundstrom L. Non-palindromic attl sites of integrons are capable of site-specific recombination with one another and with secondary targets. Mol Microbiol. 1997;26(3):441–453.
- Demarre G, Frumerie C, Gopaul DN, et al. Identification of key structural determinants of the IntI1 integron integrase that influence attC x attI1 recombination efficiency. Nucleic Acids Res. 2007;35(19):6475–6489.
- Klemm M, Cheng C, Cassell G, et al. Peptide inhibitors of DNA cleavage by tyrosine recombinases and topoisomerases. J Mol Biol. 2000;299(5):1203–1216.
- Merkel G, Andrake MD, Ramcharan J, et al. Oligonucleotide-based assays for integrase activity. Methods. 2009;47(4):243–248.
- Nakanishi-Matsui M, Hayashi Y, Kitamura Y, et al. Integrated hepatitis B virus DNA preserves the binding sequence of transcription factor Yin and Yang 1 at the virus-cell junction. J Virol. 2000;74(12):5562–5568.
- Bouvier M, Demarre G, Mazel D. Integron cassette insertion: a recombination process involving a folded single strand substrate. EMBO J. 2005;24(24):4356–4367.
- Cagle CA, Shearer JE, Summers AO. Regulation of the integrase and cassette promoters of the class 1 integron by nucleoid-associated proteins. Microbiology. 2011;157(10):2841–2853.
- Yan Z, Bryant KF, Gregory SM, et al. HIV integrase inhibitors block replication of alpha-, beta-, and gammaherpesviruses. mBio. 2014;5(4):e01318–14.
- Behdani M, Zeinali S, Karimipour M, et al. Expression, purification, and characterization of a diabody against the most important angiogenesis cell receptor: vascular endothelial growth factor receptor 2. Adv Biomed Res. 2012;1:34.
- Collis CM, Hall RM. Site-specific deletion and rearrangement of integron insert genes catalyzed by the integron DNA integrase. J Bacteriol. 1992;174(5):1574–1585.
- Rowe-Magnus DA, Mazel D. The role of integrons in antibiotic resistance gene capture. Int J Med Microbiol. 2002;292(2):115–125.
- de Groot NS, Ventura S. Effect of temperature on protein quality in bacterial inclusion bodies. FEBS Lett. 2006;580(27):6471–6476.
- Jakoby WB, editor. Enzyme purification and related techniques. Vol. 104, Methods in enzymology. New York: Academic Press; 1984.
- Scopes RK. Enzyme activity and assays. In: eLS. Chichester: John Wiley & Sons Ltd; 2002.
- Francia MV, Zabala JC, de la Cruz F, et al. The IntI1 integron integrase preferentially binds single-stranded DNA of the attC site. J Bacteriol. 1999;181(21):6844–6849.
- Gravel A, Fournier B, Roy PH. DNA complexes obtained with the integron integrase IntI1 at the attI1 site. Nucleic Acids Res. 1998;26(19):4347–4355.