
ABSTRACT
Excavation of resistance genes is one of the most effective and environment-friendly measures to control the devastating rice disease caused by Magnaporthe oryzae. Many resistance genes have been mapped and characterized in the last century. Nevertheless, only a few of the total resistance genes could be really applied in the rice breeding program. Huazhan (HZ) is a new native rice restorer line developed in China and widely used in hybrid rice in recent years. HZ and its crossed combinations usually show a broad spectrum of resistance against rice blast in different rice ecosystems in China. Dissection of the genetic background of HZ is very useful for its further application. In this study, a combined method based on bulked segregation analysis (BSA) and specific length amplified fragment sequencing (SLAF-seq) was used to identify blast resistance gene(s) in HZ. A total of 56,187 SLAFs labels were captured and 9051 polymorphic SLAFs markers were analysed and procured in this study. One trait associated with candidate resistance genes region on chromosome 12 overlapping 10.2–17.6 Mb has been identified, in which 10 NBS-LRR (nucleotide-binding site-leucine-rich repeat) coding genes were used as resistance gene candidates. Our result indicated that SLAF-seq with BSA is a rapid and effective method for initial identification of blast resistance genes. The identification of resistance gene in HZ will improve its molecular breeding and resistance variety application.
Introduction
Many genetic markers including morphological markers, biochemical markers and DNA markers such as RFLP, RAPD, AFLP, STS, SSR, CAPS, etc. have been used to identify different qualitative genes and quantitative trait locus (QTL).[Citation1–3] DNA markers based on different polymorphism-detecting techniques have promoted the gene mapping and marker-assisted selection (MAS) breeding.[Citation4] Among the genetic markers, Single nucleotide polymorphisms (SNPs) are more serviceable than other conventional markers because of its abundance and stability.[Citation5] Utilization efficiency of DNA markers needs to improve since most of the classical DNA markers are ordinarily used as single locus to map qualitative loci. High-throughput sequencing technologies offer more efficient methods for sequence-based SNP genotyping, which is more helpful for genetic mapping and genome analysis.[Citation6] Combination of specific length amplified fragment sequencing (SLAF-seq) and reduced representation library (RRL) construction has provided an efficient method for large-scale de novo SNP discovery and genotyping.[Citation7] For example, Chen et al. (2013) developed 89 SLAF markers in Thinopyrum elongatum.[Citation8] Xia et al. (2015) identified a new maize inflorescence meristem mutant based on association analysis using the SLAF-seq method.[Citation9] Bulked segregant analysis (BSA) method was considered to be an elegant method of mapping resistance genes.[Citation10,Citation11] Many blast resistance genes such as Pi37, Pi39, Pi41, Pi50, Pi58 and Pi61, etc. were mapped by this traditional method.[Citation12–17] However, the availability of molecular markers limits gene fine mapping in some cases, because of the poor polymorphism among similar genetic background. Furthermore, genotyping of each marker in the whole population is still costly and time-consuming. Whole genome sequencing of individuals pools (Pool-seq) provides a cost-effective alternative method for gene identification.[Citation18] Pool-seq method has been increasingly used for population genomic research on both model and non-model organisms.[Citation19–21] The Pool-seq technique has also been applied for rice blast resistance QTL mapping.[Citation22]
Rice blast caused by the ascomycete fungus Magnaporthe oryzae could infect the leaves, leaf collars, panicles, culms and culm nodes, which is known to occur in more than 85 countries worldwide.[Citation23,Citation24] It is estimated that destroyed rice by blast could be enough feed for more than 60 million people each year.[Citation25] Blast disease has never been eradicated from a region around the world. In order to control blast disease effectively, an integrated management program was ordinarily implemented to avoid overuse of a single control method. Application of resistant varieties is always considered to be one of the most effective and environmental methods to control blast disease, since chemical control such as carpropamid-type fungicide could pollute the ecosystem.[Citation26,Citation27] Between the qualitative and quantitative blast resistance, QTLs are usually deemed to be more durable than qualitative resistance genes, but the qualitative resistance genes were utilized more easily than that of quantitative resistance in rice breeding,[Citation28] thus breeders are inclined to exploit resistance genes in the rice breeding program.
Although more than 100 blast resistance genes have been identified or mapped based on genetic or molecular marker method [Citation28–31] and 27 of which have already been characterized,[Citation32–34] only a few resistance genes such as Pi2, Pi9, Pi39, Pi40, Pi50 and Pikh, etc. have been reported to possess broad-spectrum resistance,[Citation35–38] while most of them confer race specific resistance including Pia, Pib, Pii, Pikm, Pit, Pi12 and Pi19, etc.[Citation38,Citation39] It is still indispensable to excavate more durable resistance genes from more germplasm, since blast resistance would often break down for the reason that rare pathotypes increase or novel pathotypes emerge.[Citation40–42] In this study, a rice blast resistance gene derived from the restorer line Huazhan (HZ) was identified and mapped based on SLAF-seq method.
Materials and methods
Plant material
HZ is a new restorer line developed in China, which has been used widely in recent years. The hybrid rice combinations derived from HZ show high blast resistance in China. An F2 population with 219 individuals derived from Lijiangxintuanheigu (LTH, susceptible parent) and HZ was used for genetic analysis and resistance gene mapping. All the seeds including two parents and their corresponding 8 F1 and 219 F2 plants were evenly sown in porcelain trays (30 × 20 × 5 cm) filled with yellow mud in greenhouse. Soil was kept moist with water by auto-spraying equipment. Plants were fertilized with 3–5 g of (NH4)2SO4/m2 before inoculation at 3–4 leaf stage.
Fungal material
The M. oryzae isolate GD10-3001 which was collected from South China in 2010 showed stable pathogenicity. GD10-3001 was separated from the original strain through single spore separation method and then conserved on sterilized rice stems in a refrigerator. The isolate was transferred to yeast culture and incubated in a biochemical incubator at 25 °C for 7–10 days referred to Zhu et al.[Citation15] Then the mycelium was shifted to sterilized maize grains and cultured under purple light at 25 °C for 10–13 days. The isolate sticked on maize was transferred to sterilized porcelain plates (25 cm × 19 cm × 2 cm) covered with a piece of wet gauze and incubated at 25 °C for 3–4 days. The conidium was washed from the maize by sterilized water referred to Chen et al.[Citation29] The concentration of spores was adjusted to 1 × 105/mL mixed with 0.2% of Tween-20 before inoculation.
Rice pathogen inoculation and resistance evaluation
Rice pathogen inoculation was performed as described previously.[Citation15] Seven days later, blast symptoms were evaluated using a 0–9 scale standard based on the lesion types described by Standard Evaluation System for rice (SES, International Rice Research Institute (IRRI) 1996). Scores identified as 0–3 were considered to be resistant, scores with 4–9 were considered as susceptible.
Rice DNA extraction
Total genomic DNA was extracted from young leaves of F2 population and their parents according to the cetyltrimethyl ammonium bromide (CTAB) method.[Citation43] The total DNA was detected with NanoDrop ND-1000 Spectrophotometer (NanoDrop Technology, Wilmington, DE, USA) and diluted to 50 ng/ul. RNase A was used to remove RNA contamination from genomic DNA.
SLAF library construction and high-throughput sequencing
The reference genome of japonica rice Nipponbare, with the updated version build 5.0, was downloaded (http://rgp.dna.affrc.go.jp/IRGSP/Build3/build3.html). A pre-design SLAF experiment was performed to build up conditions to maximize SLAF-seq efficiency. First, we estimated the restriction lengths of fragments using the drilling data derived from the rice reference genome. The criteria were consulted as followed: suitable SLAFs numbers for gene mapping, average distribution of SLAFs and avoidance of repeated SLAFs. To ensure the sequencing depth consistency, a tight length range of about 30–50 bp was selected. A pilot polymerase chain reaction (PCR) amplification was performed to detect the RRL features in target length range. The pre-design step of the scheme was adjusted when non-specific amplified bands detected on the gel. The pilot experiment was to assure that the SLAFs were evenly distributed across the rice genome. The SLAF library was constructed according to the optimized pre-designed scheme. The procedure was carried out based on Sun et al. (2013) with minor adjustments.[Citation7] All of the purified DNA samples were incubated at 37 °C with MseI (New England Biolabs, NEB), T4 DNA ligase (NEB), ATP (NEB) and MseI adapter. Restriction-ligation reactions were heat-inactivated at 65 °C first, and then the samples were digested with another restriction enzyme EcoRI at 37 °C. PCR reaction was performed with mixtures of diluted restriction-ligation samples, dNTP, MseI primer containing barcode1 and Taq DNA polymerase (NEB). PCR products were purified by an E.Z.N.A.® Cycle Pure Kit (Omega) before pooled. All the purified products were run on a 2% agarose gel. Fragments of about 330–380 bp in size were isolated using a Gel Extraction Kit (Qiagen, Hilden, Germany). The fragment products were subjected to PCR reaction with Phusion Master Mix (NEB) and Solexa amplification primer mix before adding barcode2. The target products were gel-purified and diluted for pair-end sequencing through an Illumina high-throughput sequencing platform (Illumina, Inc. San Diego, CA, USA).
SLAF-seq data grouping and genotype definition
In sequencing data assay, P, M, aa and ab value represented resistant parent HZ, susceptible parent LTH, resistant pool (Rp) and susceptible pool (Sp), respectively. All SLAF single-end reads were grouped based on sequence similarity as detected by BLAT (-tile size = 11, -step size = 11).[Citation44] Sequences with 90% or more similarity were clustered into one SLAF locus. Alleles were specified to each SLAF based on the minor allele frequency evaluation. Four kinds of SLAF tags are in one locus, since rice is a diploid species. Repetitive SLAFs were filtered out. SLAFs with sequence depth less than 213 were considered as low-depth SLAFs, which were unsuited for further analysis. SLAFs with two, three or four tags were considered to polymorphic SLAFs and regarded as potential markers for further polymorphic analysis. The potential markers were classified into three segregation patterns. We confirmed the origin of alleles according to sequencing depth of the parents and R/S pools. The potential markers with one genotype deriving from P and the other from M were verified as polymorphic markers.
Resistance gene association analysis
SLAF markers with Ratio_ab ≥ 2 were considered to be Diff_markers. Regions with three or more consecutive Diff_markers were regarded as candidate resistance gene(s) regions. Difference ratio was counted as follows: Paa represented depth of Rp (aa) population derived from P, and Maa indicated depth of Rp (aa) population derived from M; Pab represented depth of Sp (ab) population derived from P, and Mab referred to depth of Sp (ab) population derived from M. The associated value was calculated as follows: Ratio_aa = Paa/Maa; Ratio_ab = Mab/Pab. The Diff_markers’ statistics procedure was as follows: two kinds of genotypes derived from parents were selected according to suitable depth (60–300) after standardizing the parents’ markers data. Diff_markers were screened according to the difference values of Rp and Sp. The gene mapping candidate regions were ascertained based on the distribution and difference ratio of Diff_markers.
Resistance spectrum analysis of HZ and eight near isogenic lines (NILs)
A set of monogenic lines carrying different major single blast R genes, kindly provided by IRRI, including four broad spectrum major genes Pi2,Pii,Pi5,Pi7 and four blast resistance genes Pita,Pita-2,Pi12,Pi19 located on chromosome 12, were used as the monogenic lines. Fifty M. oryzae representative isolates collected from South China were used to evaluate the blast resistance spectrum (RS) of restorer line HZ carrying Pi-hz(t) gene and the monogenic lines mentioned above (Table S2). RS was calculated as followed fomula: RS = (number of incompatible isolates/number of total tested isolates) × 100%.
Results and discussion
Genetic analysis of resistance gene and construction of gene pools
The resistant donor HZ was crossed with susceptible parent LTH. Their F1 individuals all showed resistant phenotype against M. oryzae isolate GD10-3001. An F2 population derived from HZ/LTH was used to analyse resistance inheritance of HZ against GD10-3001. Segregation of resistant (R) and susceptible (S) progenies of 219 F2 individuals fitted a 3:1 ratio (171 R: 48 S, x2 = 0.42, P = 0.52). The segregated result suggested that HZ harbours a dominant resistance gene designated as Pi-hz(t) tentatively. The plants with disease scales 0–2 were considered to be distinctly resistant, while the plants with disease scales between 6 and 9 were recognized to be distinctly susceptible. In this study, two parents HZ and LTH, 30 distinctly resistant and 30 clearly susceptible F2 individuals DNA were extracted and mixed as Rp and Sp for further SLAF-seq and super-BSA analysis.
SLAF-sequencing and SLAF tags data analysis
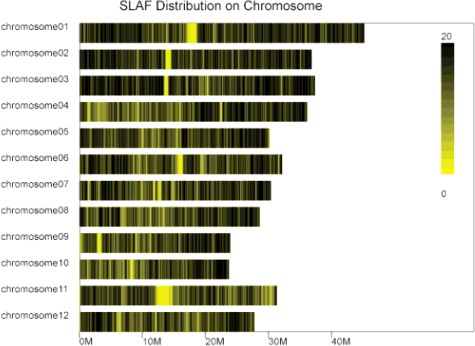
Total DNAs of four samples HZ, LTH, Rp and Sp were digested into fragments for SLAF library construction and SLAF-sequencing. After SLAF library construction and high-throughput sequencing, a total of 3,670,254,150 Mb data including 18,169,575 single-end reads were procured with each read length of ∼202 bp originally. Among them, only 1.69 Gb data including 6,190,100 valid single-end reads, which were constructed from the samples of HZ (1,121,190), LTH (1,704,652), Rp (2,050,068) and Sp (1,314,190), respectively (). The number of SLAF tags was 56,187, and the average coverage for each tag was 108.79-fold. All the SLAF tags were drew evenly in the distribution diagram on each rice chromosome (), which illustrated that the rice genome has been successfully simplified, since the SLAF tags were positioned on the rice genome equally.
Table 1. Sequencing data of each sample.
Figure 1. SLAF tag distribution on each rice chromosome.
SLAF tags polymorphic analysis
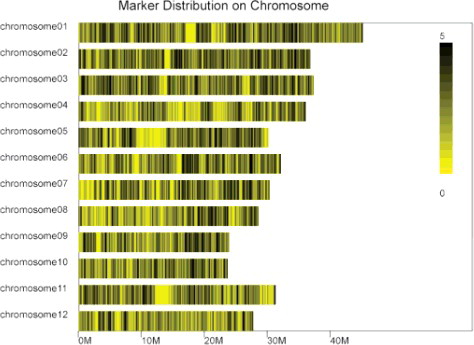
The 56,187 high-quality SLAF tags were divided into six kinds of markers. Among the SLAF markers, 9051 polymorphic markers were identified based on polymorphic analysis of allele numbers and variation among gene sequences. Among the 56,187 SLAF tags, the polymorphic tags markers were divided into three kinds: INDEL (insertion or deletion), SNP and EDP (endonuclease digestion polymorphism), of which their polymorphism rates were 0.24%, 15.62% and 0.23%, respectively. The other useless types were: no polymorphism (81.29%), unknown type (2.11%) and repeat sequence (0.48%), etc. The SLAF tags were distributed explicitly and evenly on each rice chromosome (), which proved that the SLAF-seq was prosperous.
Figure 2. Markers distribution on each rice chromosome.
Association analysis and gene annotation of the target region
The genetic analysis of the F2 population indicated that HZ contained at least a dominant resistance gene. The F2 population of HZ/LTH was divided into distinctly resistant and susceptible pools with 30 individuals each. We simplified the genetic model for association analysis. A total of 5648 markers with one genotype deriving from P and the other from M were selected to compute the difference ratio of Rp and Sp. 91 Diff_markers were obtained and mapped on chromosome 12 according to association analysis. The Diff_markers distributed on chromosome 12 were visualized in statistically. The associated assay indicated that target gene was mapped on 10M–20M on chromosome 12, since this region had clear concentration of Diff_markers based on the Ratio_ab above green lines and Ratio_aa under purple lines (). Further signal amplification of this region narrowed the target gene in 10M–18M interval (), since the Diff_markers were mapped on this region. The intervening genomic region (about 7.4 Mb) of chromosome 12, where the target gene was mapped, contains at least 143 genes annotated according to the International Rice Genome Sequencing Project (IRGSP) build 5.0 (; supplementary Table S1; ).
Figure 3. Ratio distribution of Diff_markers on chromosome 12. The Ratio_ab above the green line and Ratio_aa under the purple line indicated that corresponding markers are linked with target gene. There was a obvious concentrated region on 10M–20M based on the Diff_marker above the green and under the purple line.
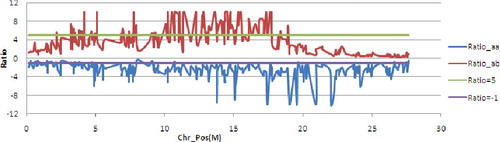
Figure 4. Ratio distribution of Diff_markers on 10M–18M of chromosome 12. The 91 Diff_markers on chromosome 12 were selected to carry out association analysis. The regions with three or more consecutive Diff_markers were considered to be related candidate gene regions. Nine related candidate gene regions overlapped 10.2M–17.6M on chromosome 12 were obtained and there were 42 Diff_markers and 143 annotated candidate genes in which based on the conference genome sequence version IRGSP build 5.0 (). Taken together, the candidate gene regions of the Pi-hz(t) gene covered a size of 7.4 Mb, approximately.
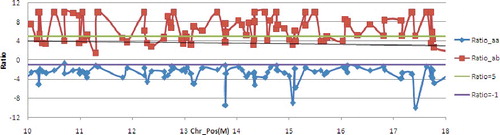
Table 2. Information of the association region.
Resistance characterization of HZ and eight NILs
Resistance evaluation of HZ and eight NILs against 50 M. oryzae isolates collected from South China indicated that HZ conferred a high RS with 94% of tested isolates, the NIL IRBLz5-CA had higher RS with 90%, two NILs IRBL7-M and IRBLta2-Pi had middle RS with 58% and 46%, and five NILs IRBL5-M, IRBLi-F5, IRBLta-CP1, IRBL12-M and IRBL19-A had lower RS with 28%, 26%, 12%, 6% and 6%, respectively (; Table S2). The resistance characterization of HZ and eight NILs against M. oryzae isolates derived from South China suggested that HZ can be used as an excellent resistant donor for MAS breeding in South China.
Table 3. Resistance spectrum of HZ and eight near isogenic lines against 50 M. oryzae isolates from South China.
Conclusions
In this study, 56,187 of SLAF tags had been developed with SLAF method. Nine thousand and fifty-one polymorphic markers were identified with high quality and quantity that met the requirements. The integral and accurate SLAF tags were well-distributed on each rice chromosome. The 91 Diff_markers provided useful data for precisely narrowing down the candidate trait-associated regions into a 7.4 Mb interval which had 143 genes annotated by IRGSP build 5.0. Annotation of the 143 candidate genes showed that there are 10 NBS-LRR (nucleotide-binding site-leucine-rich repeat) coding genes as resistant candidates in the target region, which could contribute to isolate the Pi-hz(t) gene in further research. BSA combined with SLAF-seq method could be an efficient and cost-effective way for resistance genes or QTLs mapping. Resistance characterization analysis of HZ and eight NILs showed that HZ and IRBLz5-CA had higher RS with 94% and 90%, which suggested that HZ and IRBLz5-CA would be excellent resistance source for improving blast resistance breeding program in South China. The lower level RS of NILs IRBL5-M, IRBLi-F5, IRBLta-CP1, IRBL12-M and IRBL19-A indicated that resistance genes Pi5, Pii, Pita, Pi12 and Pi19 are not recommended to apply in South China.
Acknowledgments
The authors greatly appreciate the kind help of Biomarker Technologies Company Limited in providing the technology support.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Montaldo HH, Meza-Herrera CA. Use of molecular markers and major genes in the genetic improvement of livestock. Electron J Biotechnol. 1998;2:83–89.
- Collard BCY, Mackill DJ. Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos Trans R Soc Lond B Biol Sci. 2008;363:557–572.
- Tan C, Han Z, Yu H, et al. QTL scanning for rice yield using a whole genome SNP array. J Genet Genomics. 2013;40:629–638.
- Collard BCY, Jahufer MZZ, Brouwer JB, et al. An introduction to markers, quantitative trait loci (QTL) mapping and marker-assisted selection for crop improvement: the basic concepts. Euphytica. 2005;142:169–196.
- Liu J, Huang S, Sun M, et al. An improved allele-specific PCR primer design method for SNP marker analysis and its application. Plant Methods. 2012;8:34.
- Huang X, Feng Q, Qian Q, et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009;19:1068–1076.
- Sun X, Liu D, Zhang X, et al. SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS One. 2013;8:e58700.
- Chen S, Huang Z, Dai Y, et al. The development of 7E chromosome-specific molecular markers for Thinopyrum elongatum based on SLAF-seq technology. PLoS One. 2013;8:e65122.
- Xia C, Chen L, Rong T, et al. Identification of a new maize inflorescence meristem mutant and association analysis using SLAF-seq method. Euphytica. 2015;202:35–44.
- Giovannoni JJ, Wing RA, Ganal MW, et al. Isolation of molecular markers from specific chromosome intervals using DNA pools from existing mapping populations. Nucleic Acids Res. 1991;19:6553–6558.
- Michelmore RW, Paran I, Kesseli RV. Identification of markers linked to disease resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc Natl Acad Sci USA. 1991;88:9828–9832.
- Chen S, Wang L, Que Z, et al. Genetic and physical mapping of Pi37(t), a new gene conferring resistance to rice blast in the famous cultivar St. No. 1. Theor Appl Genet. 2005;111:1563–1570.
- Liu X, Yang Q, Lin F, et al. Identification and fine mapping of Pi39(t), a major gene conferring the broad-spectrum resistance to Magnaporthe oryzae. Mol Genet Genomics. 2007;278:403–410.
- Yang Q, Lin F, Wang L, et al. IdentiWcation and mapping of Pi41, a major gene conferring resistance to rice blast in the Oryza sativa subsp. indica reference cultivar, 93-11. Theor Appl Genet. 2009;118:1027–1034.
- Zhu X, Chen S, Yang J, et al. The identification of Pi50(t), a new member of the rice blast resistance Pi2/Pi9 multigene family. Theor Appl Genet. 2012;124:1295–1304.
- Koide Y, Telebanco-Yanoria MJ, Fukuta Y, et al. Detection of novel blast resistance genes,Pi58(t) and Pi59(t), in a Myanmar rice landrace based on a standard differential system. Mol Breeding. 2013;32:241–252.
- Lei C, Hao K, Yang Y, et al. Identification and fine mapping of two blast resistance genes in rice cultivar 93-11. The Crop J. 2013;1:2–14.
- Schlötterer C, Tobler R, Kofler R, et al. Sequencing pools of individuals –mining genome-wide polymorphism data without big funding. Nature Rev Genet. 2014;15:749–763.
- Wenger WJ, Schwartz K, Sherlock G. Bulk segregant analysis by high-throughput sequencing reveals a novel xylose utilization gene from Saccharomyces cerevisiae. PLoS Genet. 2010;6:e1000942.
- Parts L, Cubillos FA, Warringer J, et al. Revealing the genetic structure of a trait by sequencing a population under selection. Genome Res. 2011;21:1131–1138.
- Swinnen S, Schaerlaekens K, Pais T, et al. Identification of novel causative genes determining the complex trait of high ethanol tolerance in yeast using pooled-segregant whole-genome sequence analysis. Genome Res. 2012;22:975–984.
- Takagi H, Abe A, Yoshida K, et al. QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013;74:174–183.
- Zeigler RS, Leong SA, Teng PS. Rice blast disease. Kew: CAB International Mycological Institute; 1994.
- Srivastava D, Shamim M, Kumar D, et al. Morphological and molecular characterization of Pyricularia oryzae causing blast disease in rice (Oryza sativa) from North India. Int J Sci Res Publ. 2014;4:1–9.
- Pennisi E. Armed and dangerous. Science. 2010;327:804–805.
- Sawada H, Sugihara M, Takagaki M, et al. Monitoring and characterization of Magnaporthe grisea isolates with decreased sensitivity to scytalone dehydratase inhibitors. Pest Manag Sci. 2004;60:777–785.
- Miah G, Rafii MY, Ismail MR, et al. Blast resistance in rice: a review of conventional breeding to molecular approaches. Mol Biol Rep. 2013;40:2369–2388.
- Sirithunya P, Tragoonrung S, Vanavichit A, et al. Quantitative trait loci associated with leaf and neck blast resistance in recombinant inbred line population of rice (Oryza sativa). DNA Res. 2002;9:79–88.
- Chen S, Su J, Han J, et al. Resistance spectrum assay and fine mapping of the blast resistance gene from a rice experimental line, IRBLta2-Re. Euphytica. 2014;195:209–216.
- Liu Y, Qi S, Young N, et al. Characterization of resistance genes to rice blast fungus Magnaporthe oryzae in a “Green Revolution” rice variety. Mol Breeding. 2015;35:52.
- Xiao W, Yang Q, Sun D, et al. Identification of three major R genes responsible for broad-spectrum blast resistance in an indica rice accession. Mol Breeding. 2015;35:49.
- Fukuoka S, Yamamoto S, Mizobuchi R, et al. Multiple functional polymorphisms in a single disease resistance gene in rice enhance durable resistance to blast. Sci Rep. 2014;4:4550.
- Ma J, Lei C, Xu X, et al. Pi64, encoding a novel CC-NBS-LRR protein, confers resistance to leaf and neck blast in rice. Mol Plant Microbe Interact. 2015;28:558–568.
- Su J, Wang W, Han J, et al. Functional divergence of duplicated genes results in a novel blast resistance gene Pi50 at the Pi2/9 locus. Theor Appl Genet. 2015;128:2213–2225.
- Liu G, Lu G, Zeng L, et al. Two broad-spectrum blast resistance genes, Pi9(t) and Pi2(t), are physically linked on rice chromosome 6. Mol Genet Genomics. 2002;267:472–480.
- Sharma TR, Madhav MS, Singh BK, et al. High-resolution mapping, cloning and molecular characterization of the Pi-k(h) gene of rice, which confers resistance to Magnaporthe grisea. Mol Genet Genomics. 2005;274:569–578.
- Jeung JU, Kim BR, Cho YC, et al. A novel gene, Pi40(t), linked to the DNA markers derived from NBS-LRR motifs confers broad spectrum of blast resistance in rice. Theor Appl Genet. 2007;115:1163–1177.
- Yang J, Chen S, Zeng L, et al. Race specificity of major rice blast resistance genes to Magnaporthe grisea isolates collected from indica rice in Guangdong, China. Rice Sci. 2008;15:311–318.
- Hayashi K, Yoshida H. Refunctionalization of the ancient rice blast disease resistance gene Pit by the recruitment of a retrotransposon as a promoter. Plant J. 2009;57:413–425.
- Ou SH. Pathogen variability and host resistance in rice blast disease. Ann Rev Phytopathol. 1980;18:167–187.
- Jia Y, Bryan GT, Farrall L, et al. Natural variation at the Pi-ta rice blast resistance locus. Phytopathology. 2003;93:1452–1459.
- Huang J, Si W, Deng Q, et al. Rapid evolution of avirulence genes in rice blast fungus Magnaporthe oryzae. BMC Genet. 2014;15:45.
- Murray MG, Thompson WK. Rapid isolation of high molecular-weight plant DNA. Nucl Acids Res. 1980;8:4321–4325.
- Kent WJ. BLAT – the BLAST-like alignment tool. Genome Res. 2002;12:656–664.