
ABSTRACT
Fusion expression is a promising strategy for the production of bioactive peptides in Escherichia coli. In this study, we constructed a new recombinant expression plasmid containing the coding sequence of 56-residue B1 domain of streptococcal protein G (GB1). For easy purification and cleavage of the recombinant proteins, except GB1, an engineered hexahistidine and tobacco etch virus (TEV) protease recognition sites were included in the fusion sequence. Next, we cloned the coding sequence of human epidermal growth factor (hEGF) into this new plasmid and produced the recombinant hEGF in E. coli. The bioactive hEGF is a 53-amino acid peptide and is stabilized by three intramolecular disulphide bonds. Compared with glutathione S-transferase, thioredoxin and small ubiquitin-related modifier, GB1 greatly improved the expression and solubility of hEGF. Moreover, the recombinant hEGF bound to the nickel nitrilotriacetic acid resin column, was easily cleaved by TEV protease and the free hEGF was released. The results showed that this new plasmid was appropriate for recombinant production of small bioactive peptides, such as hEGF, which contains a high proportion of hydrophobic residues and intramolecular disulphide linkages.
Introduction
The epidermal growth factor (EGF) was first isolated from mouse submaxillary gland by Cohen and his colleagues in 1962.[Citation1,Citation2] Since then, many reports have shown that EGF regulates a wide array of biological events, including cell proliferation, differentiation and migration.[Citation3–7] The bioactive human epidermal growth factor (hEGF) is a 53-amino acid peptide forming a typical EGF domain. The tertiary structure shows that this domain is composed of β-sheet and disordered sequence and is stabilized by three intramolecular disulphide bonds.[Citation8] Through binding to the epidermal growth factor receptor (EGFR) in the cell membrane, EGF can induce its autophosphorylation and initiate signal transduction, thus regulating a wide range of biological functions.[Citation9,Citation10] EGF is widely used in basic research and clinical medicine, which has created a great demand for it. Since the traditional purification is low in efficiency, it is of great importance to develop new methods to obtain hEGF on a large scale.[Citation11]
With the advent of recombinant DNA technology, fusion expression is a common strategy to produce hEGF. Glutathione S-transferase (GST), thioredoxin (Trx) and small ubiquitin-related modifier (SUMO) are the commonly used carrier proteins for fusion expression of bioactive peptides.[Citation12–14] However, due to difficulties with the correct folding of hEGF, most fusion proteins form inclusion bodies during the bacterial expression.[Citation15–17] The low expression level and poor refolding efficiency are the great challenges of the hEGF heterogeneous expression in Escherichia coli.
In this study, a new fusion partner was utilized to overcome the difficulties of the hEGF expression and purification. In this new system, hEGF is expressed fused to a B1 domain of streptococcal protein G (GB1).[Citation18] The 56-residue GB1 domain forms a compact fold that is appropriate for fusion expression of hEGF, which contains high proportion of hydrophobic residues and three intramolecular disulphide linkages. For easy purification, an extra hexahistidine (His6) and tobacco etch virus (TEV) protease recognition sites, were added in the fusion sequence.[Citation19] Compared with other fusion partners, GB1-fusion showed an improved expression level and the solubility of hEGF.
Materials and methods
Construction of GB1-fusion expression plasmid vector
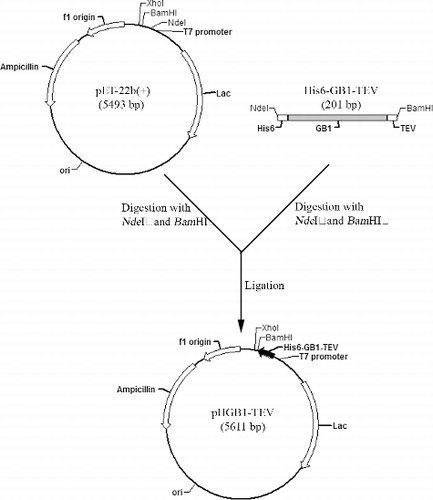
The GB1-fusion expression vector, designated as pHGB1–TEV, was constructed based on pET-22b(+) (Novagen).
The coding sequence of His6–GB1–TEV (TEV here means TEV protease recognition site) was chemically synthesized by Sangon Biotech and amplified by polymerase chain reaction (PCR) technique (MyCycle, Bio-Rad) by using the primers His6-GB1-TEV-F (Nde I) and His6-GB1-TEV-R (BamH I) (Sangon Biotech) (). The PCR reaction had a volume of 50 μL and was done in a 0.5 mL microfuge tube. The quantity of each ingredient was as follows: 800 μmol/L deoxynucleoside triphosphates (dNTP; 200 μmol/L each), 10 ng template (coding sequence of His6–GB1–TEV, ), 1 μmol/L of each primer, 1 U KOD-Plus DNA polymerase (TOYOBO), 1 × buffer (pH7.4). The temperature of the cycles was as follows: 94 °C for 1 min (denaturation), 54 °C for 1 min (annealing), 72 °C for 1 min (elongation), 30 cycles, 72 °C for 10 min at the end to allow a complete elongation of all product's DNA. The PCR products and the vector pET-22b(+) were digested with Nde I and BamH I restriction enzymes (Fermentas) following the manufacturer's instructions. Subsequently, the digested PCR fragments and the vector were purified with DNA Purification Kit (Beyotime) and ligated with a T4 ligase (TaKaRa) to form the recombinant plasmid. Next, the ligation product was transferred into the competent cells of E. coli strain Top10 by a heat shock at 42 °C for 2 min. The positive colonies, which had recombinant plasmids, were screened on an ampicillin-containing plate and the extracted plasmids were confirmed by Nde I and BamH I double digestion. Finally, the reading frame of the recombinant pHGB1–TEV plasmid was verified by DNA sequencing (BioSune).
Table 1. Synthesized coding sequences and primers used for the construction of GB1–hEGF fusion plasmid.
Clone of hEGF into different fusion expression vectors
The newly constructed pHGB1–TEV was used for the recombinant expression of GB1–hEGF. The SUMO fusion expression plasmid pET–28a–SUMO was modified from pET-28a(+) (Novagen) [Citation20] and was used to construct the SUMO–hEGF expression plasmid. Trx fusion expression plasmid, which was devoid of S-tag coding sequence, was constructed based on pET-32a(+) (Novagen) [Citation21] and was used for the construction of the Trx–hEGF expression plasmid. For the GST fusion, pGEX-4T-3 (GE Healthcare) was used. The process of cloning of hEGF into different fusion expression vectors was similar to the construction of pHGB1–TEV plasmid. Briefly, the coding sequence of hEGF (GenBank: AAS83395.1) was obtained from Sangon Biotech and amplified by PCR technique (see above) using primers hEGF-F (BamH I) and hEGF-R (Xho I) (Sangon Biotech) (). The PCR products were digested with BamH I and Xho I restriction enzymes (Fermentas) and cloned into each fusion expression plasmid. Each clone was verified by DNA sequencing (BioSune).
Solubility analysis of recombinant hEGF with different fusion proteins
Each hEGF fusion expression plasmid was transformed into E. coli strain BL21 (DE3). The solubility of each recombinant hEGF was assayed using the following procedures. Ten mL of the overnight culture was transferred into 1 L of Luria-Bertani media supplemented with 50 μg/mL kanamycin or ampicillin (Sangon Biotech) and shaken (250 rpm) at 37 °C. When A600 reached 0.6 (Beckman DU-7400 spectrophotometer), isopropyl β-D-1-thiogalactopyranoside (IPTG, Sigma-Aldrich) was added (to a final concentration of 0.2 mmol/L) for inducing the expression of hEGF fusion proteins. The cells were cultured overnight at 22 °C before harvesting. Then they were collected by centrifugation (15,000 rpm for 15 min) in a centrifugal machine Avanti J-E (Beckman), resuspended in 50 mL lysis buffer (50 mmol/L NaH2PO4, 500 mmol/L NaCl, 1% (v/v) Triton X-100, pH 8.0) and sonicated in ice bucket at 350 W for 30 min by using the JY92-IIN sonicator (Scientz). The cellular lysates were centrifuged at 15,000 rpm for 15 min to get the supernatant and the pellet fraction. The pellet fractions were dissolved with the same volume of 1% (1 g/100 mL) sodium dodecyl sulphate (SDS, Sangon Biotech) solution, as the volume of the supernatant. Equal amount of whole cell lysate, supernatant and pellet were taken out and subjected to glycine SDS-polyacrylamide gel electrophoresis (SDS-PAGE) (15% gel) followed by Coomassie Brilliant Blue R250 (Sigma) staining. The molecular weights of the proteins in the gel were indicated by the protein ladder marker (Fermentas).
Expression and purification of hEGF with GB1 fusion carrier
The expression of GB1–hEGF fusion protein and the supernatant preparation were done by using the method stated above. Next, the supernatant was passed through a Ni-nitrilotriacetic acid (Ni-NTA) agarose column (QIAGEN) with 5 mL bed volume which was equilibrated with three columns of lysis buffer (50 mmol/L NaH2PO4, 500 mmol/L NaCl, 1% Triton X-100, pH 8.0). After that, the Ni-NTA resin was thoroughly washed with wash buffer (50 mmol/L NaH2PO4, 500 mmol/L NaCl, 50 mmol/L imidazole, pH 8.0) and equilibrated with three columns of digestion buffer (50 mmol/L Tris-HCl, 0.5 mmol/L ethylenediaminetetraacetic acid (EDTA), 1 mmol/L dithiothreitol (DTT), pH 8.0). Next, 1 mg of the prepared TEV protease was added and the column was sealed and put at 22 °C overnight. After cleavage of the expressed protein, the free hEGF was eluted with three columns of digestion buffer. All the samples from each step were subjected to Tricine–SDS-PAGE (16% gel) analysis.
Mitogenic activity of recombinant hEGF
For the mitogenic activity test of hEGF, methylthiazoleterazolium (MTT) assay was used to evaluate the number of viable 3T3 cells. Mouse 3T3 fibroblast cells (NIH-3T3) were cultured in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco) and grown at 37 °C under a humidified atmosphere containing 5% CO2. When the culture reached its exponential phase, the cells were resuspended with serum-free DMEM to a density of 5 × 104 cells/mL and transferred to a 96-well plate in a final volume of 100 μL culture medium (serum-free DMEM) per well. After cultivation for 24 h, the cells were washed once with phosphate buffered saline (PBS; 140 mmol/L NaCl, 2.7 mmol/L NaCl, 50 mmol/L NaH2PO4, pH 7.4) and fresh DMEM with different concentrations of hEGF (0, 10, 20, 40, 80 ng/mL) were added. The cells were incubated at 37 °C under a humidified atmosphere containing 5% CO2 for 72 h. The number of viable cells was determined by adding MTT (5 mg/mL) to each well and incubating for another 4 h. After discarding the culture medium 100 μL dimethyl sulfoxide (DMSO) was added to each well and was shaken (60 rpm) at room temperature for 15 min. The absorbance was immediately measured by enzyme-linked immunosorbent assay (ELISA) at 570 nm on Thermo MK3 following the manufacturer's instructions.
Results and discussion
Construction of hEGF recombinant expression plasmid with different fusion carriers
We developed a new plasmid for the expression of GB1-fused hEGF. The new GB1-fusion expression plasmid, designated as pHGB1–TEV, was constructed based on pET-22b(+). For easy purification and cleavage, the engineered fusion sequence contained three elements: the central GB1 sequence, the N-terminal His6 and the C-terminal TEV protease recognition site (). The fusion sequence was inserted into pET-22b(+) with Nde I and BamH I restriction enzyme sites to form the plasmid pHGB1–TEV, which can be seen in . With this vector, the hEGF recombinant expression plasmid was constructed to express GB1–hEGF. For comparison, we constructed other hEGF recombinant expression plasmids to produce recombinant SUMO–hEGF, Trx–hEGF and GST–hEGF.
Figure 1. Engineered fusion sequence containing GB1, N-terminal hexahistidine (His6) tag and C-terminal TEV protease recognition site.

Figure 2. Construction of the pHGB1–TEV plasmid.
Because of the difficulties in the hEGF folding, the bioactive recombinant hEGF is hard to obtain by using a prokaryotic expression system, even with fusion proteins. We introduced GB1 as a better fusion carrier of hEGF and compared it with other fusion proteins. SUMO, Trx and GST are the common carrier proteins for recombinant expression of proteins with low expression level and solubility.[Citation20,Citation21] Herein, we constructed different fusion expression plasmids to study the production of recombinant hEGF in E. coli.
High expression level and solubility of hEGF with GB1 fusion
By using the constructed recombinant expression plasmids, an expression of hEGF with different fusion proteins was induced in E. coli strain BL21 (DE3). (A) demonstrates the constructs of recombinant hEGF expressed with the different fusion proteins. The Trx and GST fusions could be cleaved by thrombin, whereas the SUMO and GB1 could be removed by SUMO protease and TEV protease, respectively.
Figure 3. Recombinant hEGF with different fusion proteins: Schematics (A); arrows indicate different enzymes cleavage sites. SDS-PAGE analysis (B) of expression level and solubility of hEGF with different fusion proteins; arrows indicate each recombinant hEGF.
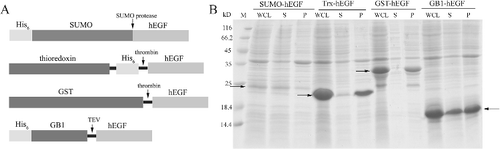
The expression levels and the solubility of the different fusion proteins were analysed by using SDS-PAGE and are shown in (B). As previously reported, SUMO ran on an SDS-PAGE gel as 20 kDa, although its calculated molecular weight is about 10 kDa.[Citation22] So, SUMO–hEGF was about 25 kDa on the gel. As we can see, the expression level of SUMO–hEGF was very low. The GST–hEGF and Trx–hEGF showed high expression levels, but low solubility. Only the GB1–hEGF fusion protein demonstrated the high expression level and high solubility. Therefore, GB1 turned out to be the most appropriate fusion carrier for the expression and purification of the recombinant hEGF from E. coli.
Expression and purification of hEGF with GB1 fusion carrier
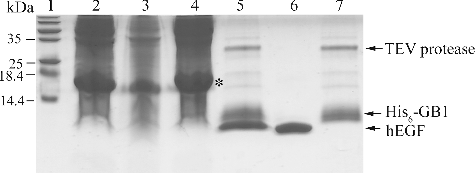
Having selected the GB1–hEGF fusion, due to its high expression level and high solubility, we performed the purification process by Ni-NTA affinity chromatography. Due to the His6 and TEV protease cleavage sites in the fusion sequence, the recombinant hEGF can be easily purified and the free hEGF can be easily separated. The TEV protease was cloned into pET-28a, expressed in E. coli strain BL21 (DE3) and purified by Ni-NTA.[Citation23,Citation24] After the efficient digestion of the fusion protein by TEV protease, the hEGF was released from the Ni-NTA column. The results can be seen in . Since both the fusion sequence and the TEV protease have His6 which can bind the Ni-NTA resin, the free hEGF was separated and eluted by the Ni-NTA affinity chromatography. The cleavage leaved only an extra glycine residue at the N-terminus of the hEGF peptide.
Figure 4. Tricine–SDS-PAGE analysis of purified hEGF.
Mitogenic activity of recombinant hEGF
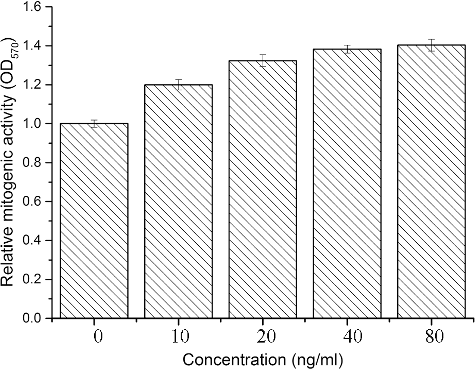
We performed a mitogenic activity test to obtain results about the biological activity of the purified hEGF. As it can be seen from , the MTT assays showed that the free hEGF had mitogenic activity in a dose-dependent manner.
Figure 5. Mitogenic activity analysis of different concentrations of hEGF to the NIH-3T3 cells in vitro.
Conclusions
Nowadays, it is easy to get the coding sequence of a target protein and clone it into an expression plasmid. So, it is a common strategy to obtain the recombinant protein using prokaryotic expression system. Because of the complexity of the protein folding and the simplicity of the prokaryotic expression system, there are a number of difficulties associated with the expression of heterologous proteins in E. coli host cells, such as protein folding and insolubility. Application of fusion expression system is a good strategy for solving these problems. In this study, we constructed a new GB1 fusion plasmid and studied the expression level and solubility of the recombinant hEGF with different fusion proteins. Compared with SUMO, Trx and GST, GB1 not only increased the expression level, but also enhanced the solubility of hEGF. With the GB1 fusion expression system, the hEGF could be easily purified and showed a high biological activity. Production of the recombinant hEGF or other bioactive proteins in E. coli with the GB1 fusion expression system is an efficient low-cost strategy and, therefore, has a great commercial value.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Cohen S. Isolation of a mouse submaxillary gland protein accelerating incisor eruption and eyelid opening in the new-born animal. J Biol Chem. 1962;237:1555–1562.
- Kleinzeller A, Martin BR, editors. Current topics in membranes and transport. Vol. 18, Membrane receptors. New York (NY): Academic Press; 1983.
- Wingren U, Franzen L, Larson GM, et al. Epidermal growth factor accelerates connective tissue wound healing in the perforated rat mesentery. J Surg Res. 1992;53:48–54.
- Cohen S, Taylor JM. Epidermal growth factor: chemical and biological characterization. Symp Soc Dev Biol. 1974;30:25–42.
- Yu FS, Yin J, Xu K, et al. Growth factors and corneal epithelial wound healing. Brain Res Bull. 2010;81:229–235.
- Schlessinger J, Schreiber AB, Levi A, et al. Regulation of cell proliferation by epidermal growth factor. CRC Crit Rev Biochem. 1983;14:93–111.
- Feng Y, Dai X, Li X, et al. EGF signalling pathway regulates colon cancer stem cell proliferation and apoptosis. Cell Prolif. 2012;45:413–419.
- Lu HS, Chai JJ, Li M, et al. Crystal structure of human epidermal growth factor and its dimerization. J Biol Chem. 2001;276:34913–34917.
- Lemmon MA, Bu Z, Ladbury JE, et al. Two EGF molecules contribute additively to stabilization of the EGFR dimer. EMBO J. 1997;16:281–294.
- Gamou S, Shimizu N. Epidermal growth factor and its receptor: the structure and function. Nihon Rinsho. 1992;50:1885–1892.
- Jaspar JM, Franchimont P. Purification of human epidermal growth factor (urogastrone) from urine. Eur J Biochem. 1987;166:295–301.
- Li Y. Carrier proteins for fusion expression of antimicrobial peptides in Escherichia coli. Biotechnol Appl Biochem. 2009;54:1–9.
- Li Y. Recombinant production of antimicrobial peptides in Escherichia coli: a review. Protein Expr Purif. 2011;80:260–267.
- Lu W, Cao P, Lei H, et al. High-level expression and purification of heparin-binding epidermal growth factor (HB-EGF) with SUMO fusion. Mol Biotechnol. 2010;44:198–203.
- Ferrer Soler L, Cedano J, Querol E, et al. Cloning, expression and purification of human epidermal growth factor using different expression systems. J Chromatogr B. 2003;788:113–123.
- Sharma K, Babu PV, Sasidhar P, et al. Recombinant human epidermal growth factor inclusion body solubilization and refolding at large scale using expanded-bed adsorption chromatography from Escherichia coli. Protein Expr Purif. 2008;60:7–14.
- Lee JY, Yoon CS, Chung IY, et al. Scale-up process for expression and renaturation of recombinant human epidermal growth factor from Escherichia coli inclusion bodies. Biotechnol Appl Biochem. 2000;31:245–248.
- Cheng Y, Patel DJ. An efficient system for small protein expression and refolding. Biochem Biophys Res Commun. 2004;317:401–405.
- Rocco CJ, Dennison KL, Klenchin VA, et al. Construction and use of new cloning vectors for the rapid isolation of recombinant proteins from Escherichia coli. Plasmid. 2008;59:231–237.
- Kuo D, Nie M, Courey AJ. SUMO as a solubility tag and in vivo cleavage of SUMO fusion proteins with Ulp1. Methods Mol Biol. 2014;1177:71–80.
- Li Y. A novel protocol for the production of recombinant LL-37 expressed as a thioredoxin fusion protein. Protein Expr Purif. 2012;81:201–210.
- Satakarni M, Curtis R. Production of recombinant peptides as fusions with SUMO. Protein Expr Purif. 2011;78:113–119.
- Wei L, Cai X, Qi Z, Rong L, et al. In vivo and in vitro characterization of TEV protease mutants. Protein Expr Purif. 2012;83:157–163.
- Tropea JE, Cherry S, Waugh DS. Expression and purification of soluble His(6)-tagged TEV protease. Methods Mol Biol. 2009;498:297–307.