
ABSTRACT
This paper describes the construction and selection of a high-producing mutant, Bacillus subtilis HB-32, with enhanced acetoin yield and productivity. The mutant was obtained by the protoplast fusion of a Bacillus subtilis mutant TH-49 (Val−) producing acetoin and Bacillus licheniformis AD-30 producing α-acetolactate decarboxylase, with the fusogen polyethylene glycol and after the regeneration and selection, etc. of the fusant. The acetoin production reached 49.64 g/L, which is an increase of 61.8% compared to that of B. subtilis strain TH-49. Random amplified polymorphic DNA analysis was performed to determine the mutagenic and protoplast fusion effects and the genomic changes in the acetoin high-producing strain compared to the parent strains at the molecular level. The constructed strain was shown to be promising for large-scale acetoin production. Future studies should focus on the application of the mutant strain in practice.
Introduction
Acetoin, an agent with a four-carbon molecule, is a buttery flavour that is widely applied in food, everyday chemicals and in the medical industry, etc. It is mainly produced by a complicated chemical synthesis process which leads to severe pollution; the material obtained is unstable and the raw material sources are limited. However, the biosynthesis process is valued due to some advantages, including abundant and renewable raw material sources, an environmentally friendly production technology, mild conditions, a high-quality product obtained, etc. Nevertheless, to the best of our knowledge, so far, there is no report on the large-scale production of acetoin via a biological processes.[Citation1–3]
In addition to the Embden–Meyerhof–Parnas (EMP pathway), two key enzymes are involved in acetoin synthesis in bacteria: α-acetolactate synthase and α-acetolactate decarboxylase (ALDC).[Citation4–6] The stimulation of ALDC activity can accelerate the transformation from α-acetolactic acid to acetoin.[Citation7–10] Based on polymerase chain reaction (PCR), the RAPD (random amplified polymorphic DNA) technique has been widely used in each field of molecular biology for its advantages, including simple operation, high sensitivity, low cost and no requirement for prior knowledge of molecular genetic information, i.e. genome sequence data, etc.[Citation10–13] In this study, RAPD analysis was used to analyse the mutagenic and protoplast fusion effects and the genomic changes in an acetoin high-producing strain and its parent strains at the molecular level.[Citation14,Citation15] The process of and results from the construction of an acetoin high-producing strain are presented in this paper.
Materials and methods
Strains
Bacillus subtilis TH-49 (Val−), with 30.69 g/L acetoin production, was maintained at the Food and Fermentation Engineering Key Lab of Shandong Province.
Bacillus licheniformis AD-30 was maintained at the Food and Fermentation Engineering Key Lab of Shandong Province, for ALDC.
Main reagents and raw materials
Lysozyme was purchased from Beijing Zhongtian Noah Enzyme Preparation Co., Ltd. (Beijing, China). Polyethylene glycol (PEG 6000) was purchased from Sinopharm Chemical Reagent Co., Ltd. Sodium dodecyl sulphate (SDS) was obtained from Qingdao Haitai Biotechnology Co., Ltd. Taq DNA polymerase, deoxyribonucleoside triphosphates (dNTPs), Protease K, ethylenediaminetetraacetic acid (EDTA), Tris and ethidium bromide (EB) were all provided by Shanghai Sangon Biotech Engineering Co., Ltd.
Medium
Plate/slants medium consisted of 10 g/L peptone, 5 g/L yeast extract, 5 g/L corn steep liquor, 5 g/L NaCl, 0.05 g/L MgSO4, and 20 g/L agarose, at pH 7.0.
High osmotic regeneration complete medium was composed of 5 g/L glucose, 3 g/L yeast extract, 10 g/L peptone, 5 g/L beef extract, 0.2 g/L MgSO4, 5 g/L NaCl and 20 g/L agarose, and hyperosmotic medium at pH 7.0.
Semi-solid high osmotic regeneration complete medium was prepared by changing the concentration of agarose (20 g/L) in the high osmotic regeneration complete medium to 10 g/L.
Preparation of major solutions
The fermentation medium was composed of 120 g/L lucose, 5 g/L yeast extract, 10 g/L corn steep liquor, 5 g/L ammonium sulphate and 0.2 g/L magnesium sulphate, at pH 7.0.
Sucrose-magnesium-maleate buffer (SMM) contained 0.5 g/L sucrose, 0.02 g/L maleic acid, 5 g/L NaCl, 0.02 g/L MgCl2·6H2O and double distilled water at pH 7.0. It was sterilized at 121 °C for 20 min.
Lysozyme solution: hypertonic buffer was added to achieve the terminal concentration of 5 mg/mL, and 0.22-μm Millipore filter was used for sterilization.
Fresh calcium phosphate solutions were prepared containing CaCl2 (1 mol/L) and KH2PO4 (0.02 mol/L). Both solutions were prepared with double distilled water. Before use, they were mixed in a 1:1 (v/v) ratio and sterilized.
PEG 6000 solution: a certain amount of PEG 6000 was weighed as required and mixed with SMM to obtain a constant volume; then, the PEG treatment solution was prepared.
Tris-EDTA (TE) buffer was obtained as follows: aliquots of 5 mL Tris-HCl buffer (1 mol/L, pH 8.0) and 1 mL EDTA (0.5 mol/L, pH 8.0) were put in a 500 mL beaker. Then, 400 mL of deionized water was added to a volume of 500 mL. After sterilization, the buffer was stored at room temperature.
Tris-acetate-EDTA (TAE) buffer was prepared as follows: aliquots of 37.2 g Na2EDTA·2H2O, 242 g Tris and 800 mL deionized water were mixed in a 1-L beaker and stirred uniformly. Acetate (57.1 mL) was added, and the mixture was stirred until homogenization. Then, deionized water was added to achieve a volume of 1 L, and the solution was stored at room temperature.
Acquisition of a high-producing acetoin fusant
Preparation of the protoplast of the parent strain
Aliquots of 10 mL cell culture broth of B. subtilis TH-49 at the middle-late stage of the logarithmic phase and B. licheniformis AD-30 were taken, respectively, and were centrifuged at 4000 g for 10 min. The cells were collected after removing the supernatant and then were washed three times with SMM. Next, SMM was added to produce cell suspensions. The viable counts were determined, and the cell concentration was adjusted to approximately 2 × 108 /mL. SMM bacterial suspension (10 mL) was transferred into a 100-mL Erlenmeyer flask and 0.4 mL of the lysozyme solution was added. The enzyme concentration in the reaction system was 0.2 mg/mL. Incubation was done in a water bath at 35 °C for about 35 min. Microscopically, more than 90% of the cells were transformed to protoplasts, and the enzymatic reaction was terminated. Aliquots of 5 mL protoplasts liquid culture obtained after the enzymolysis were centrifuged at 4000 g for 10 min. The cells were collected after removing the supernatant and then were washed three times with SMM. Afterwards, SMM was added to prepare a cell suspension. The viable counts were determined and the cell concentration was adjusted to (1.0–1.5 × 108)/mL.[Citation16]
Inactivation of protoplast of donor strain B. licheniformis AD-30
Protoplast SMM suspension of the donor strain B. licheniformis AD-30 (2 mL) was transferred into a sterile tube. The inactivation temperature was maintained at 50 °C for 90 min.
Protoplast fusion and regeneration
Aliquots of 5 mL of the prepared protoplast SMM suspension of the two parent strains were taken and mixed. The mixture was centrifuged at 4000 g for 10 min. After removing the supernatant, 9.5 mL PEG 6000 solution (35%) was added; the pH was adjusted to 8.5 and 0.2 mL of fresh calcium phosphate solution (0.02 mol/L) was also added. In the end, the solution was mixed evenly. Then, it was incubated at 34 °C for approximately 30 min and then was centrifuged at 4000 g for 10 min. Aliquots of 10 mL hypertonic mutant suspension were prepared with SMM after removing the supernatant. The hypertonic mutant suspension was diluted with SMM, appropriately. A portion of the suspension (0.5 mL) was mixed with 4.5 mL of cooled (less than 50 °C) but not solidified hypertonic complete medium. The mixture was evenly spread to the plate with the lower hypertonic complete medium and was cultured at 37 °C for 2–3 d. The colonies were counted to calculate the protoplast fusion rate.
Detection of the target mutant
The colonies in the hypertonic complete medium were numbered separately and were successively inoculated using the point of an aseptic toothpick to the corresponding numbered spots of the basic, amino acid supplemented and complete media. The media were cultured at 37 °C. The colonies in the complete medium corresponding to the colonies which could grow on the amino acid supplemented medium but failed to grow on the basic medium were inoculated onto the agar slants and cultured at 37 °C for 2 d. The acetoin production was investigated by determination of flask fermentation. The cell density was determined spectrophotometrically (721 visible spectrophotometer, Beijing Hong Yuan Xin Technology Co., Ltd.) by measurement of the optical density at 620 nm (OD620).
Experimental verification of the flask fermentation of the mutant
The acetoin production was investigated by preliminary screening and rescreening through flask fermentation of the mature strains from the agar slants.
Extraction of genomic DNA from the strains
B. subtilis suspension (1 mL) cultured for 24 h was centrifuged at 10,000 g for 1 min. TE buffer (500 μL) was added to facilitate the suspension of the precipitate. Then, lysozyme (10 μL) was added and incubated in a water bath at 37 °C for 30 min; 50 μL SDS solution (10%) and 5 μL Protease K were mixed well and incubated in a water bath at 56 °C for 3 h. Saturated phenol of an equivalent volume was sufficiently mixed with the solution described above, and the mixture was centrifuged at 12,000 g for 10 min. After the centrifugation, the saturated phenol and chloroform at a volume ratio of 1:1 were mixed with the supernatant and extracted again. The supernatant obtained in the previous step, 1/10 volume of sodium acetate (pH 5.2, 3 mol/L) and 1 μL RNase A were blended sufficiently and incubated in a water bath at 37 °C for 30 min; 2.5 volumes of ethanol were added, and the solution was held at −20 °C for 2 h. Afterwards, the solution was centrifuged at 12,000 g for 10 min and washed twice with 500 μL ethanol (70%) after removing the supernatant. The precipitate was obtained by centrifugation at 12,000 g and removing the supernatant and was dried on a super-clean worktable. Finally, the precipitate was dissolved in 50 μL TE buffer.[Citation10,Citation11]
RAPD analysis
Amplification was performed with random primers () and a template of extracted DNA from Bacillus subtilis. DNA template (10 ng), Mg2+ (2 μL, 25 mmol/L), 10 × PCR buffer (2.5 μL), Taq polymerase (5 U), random primer (1 μL, 20 μmol/L) and dNTPs (3 μL, 2.5 mmol/L) were added to the reaction system, and double distilled water was added to the total volume of 25 μL. The mixture was amplified in a PCR instrument (DTC, Xi'an Tian Long Technology Co., Ltd.).
Table 1. Random primers used in this study.
The following reaction programme was used: (1) the first step was pre-denaturation at 94°C for 4 min. (2) The second step consisted of 35 cycles, each with denaturation at 94 °C for 1 min, annealing at appropriate temperature (30, 35 or 40 °C) for 1 min and extension at 72 °C for 2 min. (3) The third step was extension at 72 °C for 10 min.
After the PCR amplification, 10 μL of amplified products was analysed via TAE agarose gel electrophoresis (1% agarose, 100 V) stained with EB for 20 min and observed and photographed under an ultraviolet lamp.
Results and discussion
Screening for a target high-producing acetoin mutant
The protoplast fusion was performed under optimum fusion conditions determined in preliminary experiments. A total of 256 single colonies were selected after cultivation in double-layer regeneration hypertonic medium and mutant examination. After cultivation at a constant temperature of 37 °C for 2 d, the acetoin production was determined through flask fermentation. In a procedure of preliminary screening, 13 strains with acetoin production that was higher than that of the parent B. subtilis strain HT-49 were selected. The rescreening results are presented in
Table 2. Rescreening results from flask fermentation.
As shown in , the mutant with the highest acetoin yield was B. subtilis HB-32. Its major carbon source was 120 g/L of initial glucose. By flask fermentation, the acetoin concentration and the productivity of this mutant strain reached 49.64 g/L and 0.69 g/(L h), respectively, which were both 61.8% higher in comparison with those observed for the parent strain B. subtilis TH-49.
Further, the conversion rate of glucose was 45.5%. The acetoin production of B. subtilis HB-32 was maintained at about 49.5 g/L after multiple cycles of natural division of B. subtilis HB-32. Further research on the activity changes of key enzymes related to acetoin metabolism and the features of mutant metabolism needs to be performed.
Verification of the genetic stability and genetic marker(s) of the fusant
To further explore the genetic stability and genetic markers of the mutant B. subtilis HB-32, its growth was studied in the seventh subculture in the basic and amino acid supplemented medium. The genetic marker of the mutant B. subtilis HB-32 (Val−) was stable. The effect of cell passage and storage time at 4 °C in a refrigerator on the acetoin production of the mutant B. subtilis HB-32 was studied by flask fermentation. The results showed that acetoin production by the mutant B. subtilis HB-32 was maintained above 49.5 g/L after seven generations (), demonstrating that the high acetoin production was genetically stable in this strain.
Table 3. Stability of the mutant HB-32.
The acetoin production of the mutant B. subtilis HB-32 was also shown to be maintained above 49.5 g/L when the strain was stored at 4 °C in a refrigerator for 80 d (). The production was 49.36 g/L and decreased by only 0.28% when the strain was stored at 4 °C for 100 d. However, when it was stored for 120 d, the production decreased by 0.98%. Thus, the results indicated that the acetoin production of the mutant B. subtilis HB-32 was stable if the storage time was within 100 d, further illustrating that the constructed mutant B. subtilis HB-32 was genetically stable and could be utilized for industrial applications.
Table 4. Influence of preservation time at 4 °C in a refrigerator on the acetoin production by the mutant B. subtilis HB-32.
RAPD analysis of the parent strains and the mutant B. subtilis HB-32
The high-producing mutant B. subtilis HB-32 was selected for further investigation in this study as a potential novel strain with enhanced ALDC activity and improved production of acetoin. RAPD analysis [Citation11,Citation12] was used to analyse the mutagenic and protoplast fusion effects and the genomic changes in the acetoin high-producing strain B. subtilis HB-32 and its parent strains, the acetoin-producing mutant B. subtilis TH-49 (Val−) and the ALDC-producing B. licheniformis AD-30, at the molecular level.
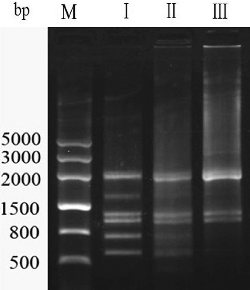
Preliminary experiments were first performed to determine the optimal PCR amplification conditions prior to RAPD analysis. Of the three different annealing temperatures used (30, 35 and 40 °C), sharp RAPD bands and a clear background were obtained when the annealing temperature was 35 °C (). The bands were not obvious and the background was obscure at 30 and 40 °C. Thus, 35 °C was set as the annealing temperature for RAPD.
Figure 1. Electrophoretogram of PCR products amplified using different annealing temperatures.
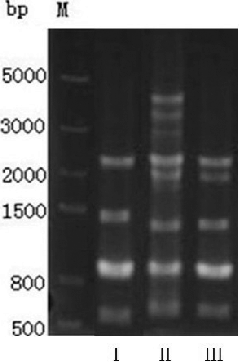
In our RAPD analysis, 20 random primers were used. Only five of these primers generated amplification products visualized as electrophoretic bands. The only primer that generated polymorphic bands was the 5'-ACCTTCGGAC-3' primer. The amplified polymorphic bands of the parent strains and the mutant are depicted in . The fusant and B. licheniformis AD-30 shared one additional band of approximately 2000 bp, which was not present in B. subtilis TH-49. This indicates that the genome of the fusant B. subtilis HB-32 contained some altered sequences in comparison to those of its parents, B. subtilis TH-49 and B. licheniformis AD-30.
Figure 2. Amplification results of strain TH-49, AD-30 and HB-32.
It is well known that the genes involved in metabolic pathways in bacteria are often organized in operons. For example, much attention has been paid to the mechanisms of gene regulation in the catabolism of acetoin and it is now known that acetoin catabolism is catalysed by the acetoin dehydrogenase system encoded by a sequence of genes linearly arranged on the chromosome with a common promoter (the acoABCL operon).[Citation17–19] Thus, by analogy, it might be speculated that, in our study, the increased acetoin production of the fusant B. subtilis HB-32 might possibly be attributed to an increased number of copies of the ALDC gene. The obtained strain could be considered promising for larger-scale production of acetoin. Further research, however, is needed to more precisely identify the underlying mechanisms of enhanced acetoin production by this strain.
Conclusions
An acetoin high-producing strain B. subtilis HB-32 was obtained from the parent strains B. subtilis TH-49 (Val−) and B. licheniformis AD-30 by the inactivated protoplast fusion technique and selection based on valine auxotrophy and flask fermentation. By flask fermentation for 72 h in a growth medium containing 120 g/L of glucose as an initial carbon source, the mutant B. subtilis HB-32 was able to produce 49.64 g/L of acetoin, with a productivity of 0.69 g/L/h. This yield exceeded that of the parent strains B. subtilis TH-49 and is, to the best of our knowledge, the highest yield currently reported in China. The mutant strain proved to be genetically and metabolically stable, preserving its RAPD genetic marker and a stable high acetoin yield for seven generations and storage at 4 °C in a refrigerator for 100 d. The results from the RAPD analysis indicated that the genome of the mutant B. subtilis HB-32 was changed to some degree compared to its parent strains. It might be speculated that the improvement of acetoin production in B. subtilis HB-32 may result from an increase in the copy number of the ALDC gene. Further research, however, is needed to better explore this hypothesis and the future perspective for application of the mutant strain in practice.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Xiao Z, Lu JR. Strategies for enhancing fermentative production of acetoin: a review. Biotechnol Adv. 2014;32:492–503.
- Tian Y, Fan Y, Zhao X, et al. Optimization of fermentation medium for acetoin production by Bacillus subtilis SF4-3 using statistical methods. Prep Biochem Biotechnol. 2014;44:529–543.
- Zhang Y, Li S, Liu L, et al. Acetoin production enhanced by manipulating carbon flux in a newly isolated Bacillus amyloliquefaciens. Bioresour Technol. 2013;130:256–260.
- Davies ME. Acetolactate and acetoin synthesis in ripening peas. Plant Physiol. 1964;39:53–59.
- Juni E. Mechanism of the formation of acetoin by yeast and mammalian tissue. J Biol Chem. 1951;195:727–734.
- Junie E. Mechanisms of formation of acetoin by bacteria. J Biol Chem. 1952;195:715–726.
- Gunashree BS, Venkateswaran G. Enhanced phytase production through interspecific protoplast fusion of Aspergillus niger CFR 335 and Aspergillus ficuum SGA 01 auxotrophic mutants. Enzyme Microb Tech. 2010;46:562–567.
- Willams JGK, Kubelik AR, Livak J, et al. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res. 1990;18:6531–6535.
- Welsh J, Mcclelland M. Fingerprinting genomes using PCR with arbitrary primers. Nucleic Acids Res. 1990;18:7213–7218.
- Leo AK, Halvorson HO. Metabolism of poly-β-hydroxybutyrate and acetoin in Bacillus cereust. J Bacteriol. 1965;90:1251–1259.
- Yang LJ, Wang JH. Genetic manipulation of the pathway for diacetyl metabolism in Lactococcus lactis. Appl Environ Microbiol. 1996;62:2641–2643.
- Andrighetto C, Psomas E, Tzanetakis N, et al. Randomly amplified polymorphic DNA (RAPD) PCR for the identification of yeasts isolated from dairy products. Lett Appl Microbiol. 2000;30:5–9.
- Williams OB, Morrow MB. The bacterial destruction of acetyl-methylcarbinol. J Bacteriol. 1928;16:43–48.
- Hugenholtz J, Kleerebezem M, Starrenburg M, et al. Lactococcus lactis as a cell factory for high-level diacetyl production. Appl Environ Microbiol. 2000;66:4112–4114.
- Olsen F, Aunstrup K. α-Acetolactate decarboxylase enzyme and preparation thereof. United States Patent US 4,617,273. 1986 Oct 14.
- Freese E, Fortnagel U. Growth and sporulation of Bacillus subtilis mutants blocked in the pyruvate dehydrogenase complex. J Bacteriol. 1969;99:745–756.
- Huang M, Oppermann FB, Steinbuchel A. Biochemical and molecular characterization of the Bacillus subtilis acetoin catabolic pathway. J Bacteriol. 1999;181(12):3837–3841.
- Oppermann FB, Schmidt B, Steinbuchel A. Purification and characterization of acetoin: 2,6-dichlorophenolindophenol oxidoreductase, dihydrolipoamide dehydrogenase and dihydrolipoamide acetyltransferase of the Pelobacter carbinolicus acetoin dehydrogenase enzyme system. J Bacteriol. 1991;173(2):757–767.
- Oppermann FB, Steinbuchel A. Identification and molecular characterization of the aco genes encoding the Pelobacter carbinolicus acetoin dehydrogenase enzyme system. J Bacteriol. 1994;176(2):469–485.