
ABSTRACT
Colorectal cancer (CRC) is the third most common cancer in the world and its disease-specific mortality is estimated to be approximately 33% in the developed world. KRAS mutations have been shown to predict response to anti-EGFR (epidermal growth factor receptor) targeted monoclonal antibody therapy. Therefore, KRAS mutation testing of metastatic CRC patients is mandatory in the clinical setting to aid in the choice of appropriate therapy. Currently, the most common strategy for KRAS mutation detection consists of conventional polymerase chain reaction (PCR) and direct sequencing. However, it is a time-consuming and complicated procedure, not suitable for routine clinical test. The objective of this study is to develop and evaluate a highly sensitive and rapid method using peptide nucleic acid (PNA) oligomers mediated real-time PCR clamping for detection of KRAS mutations. The PNA-mediated PCR clamping assay can real-time detect a mutation in a sample containing 1% of the mutant allele in a mixture of wild-type genomic DNA, which also enables the accurate and rapid detection of all KRAS codon 12 and 13 mutations in a single reaction. The total assay time is short as it requires only 1.5 hours after the samples preparation. Thus, the present method offers a potential alternative to be applied in clinical samples of CRC for detection of DNA carrying KRAS mutations.
Introduction
Colorectal cancer (CRC) is one of the most common cancers worldwide. According to WHO statistics, there were estimated over 1.2 million new CRC cases and 608,700 deaths in 2008 worldwide [Citation1]. KRAS (V-Ki-ras2 Kirsten rat sarcoma viral oncogene), encoding a 21 KDa GTP-binding protein in the epidermal growth factor receptor (EGFR) signalling pathway, can acquire activating mutations in codons 12 and 13 of exon 2 in approximately 35%–45% of the CRC cases, rendering EGFR inhibitors ineffective [Citation2]. Because KRAS mutations have been explored as prognostic biomarkers, it is considered a promising predictive for clinical decisions and anti-EGFR therapy [Citation3]. Therefore, in modern clinical settings, KRAS mutation detection has become essential for offering the most suitable therapy for a patient.
The historical standard for KRAS mutation testing has been direct sequencing of DNA extracted from samples of tumour or from biopsies [Citation4]. The advantage of this method is that it can be applied to discover unknown mutations. However, direct sequencing is too complex, time-consuming and expensive for routine pre-therapeutic screening programmes. Furthermore, the clinical samples often contain a subpopulation of mutant cells mixed within an excess of normal tissue, causing the mutations to be missed by sequencing due to technical limitations of the direct sequencing [Citation5].
Over the past decade, based on findings that the most common KRAS mutations are seven types mutant of codons 12, 13 (exon 2) and in rare cases at codon 61 (exon 3), more mutation specific approaches have been developed. These methods, including the polymerase chain reaction–restriction fragment length polymorphism (PCR-RFLP), single-strand conformation polymorphism (PCR-SSCP), mutant–allele-specific amplification and mutation enrichment assays [Citation6], high-resolution melting (HRM) analysis [Citation7] and other methodologies [Citation8]. Some methods have been proven to be more accurate and sensitive than previous direct sequencing, but most of them are still multiplex steps and/or time-consuming, highly costly and unsuitable for routine clinical screening and resource-limited settings [Citation9–Citation11]. Meanwhile, detection of a KRAS mutation in a CRC patient requires that a very sensitive assay should be used because the level of mutant-type KRAS DNA will be lower than that of wild-type KRAS DNA.
Various combinations of allele-specific PCR with real-time PCR have also been shown to effectively enrich minority alleles with moderate to high selectivity [Citation12–Citation14]. Oligonucleotides of modified molecular such as peptide nucleic acid (PNA)-based primers have also been proven to be effective for the enrichment of allele specificity in PCR screening [Citation15]. PNA is a synthetic DNA analogue, in which the phosphodiester backbone is replaced by a peptide-like repeat formed by (2-aminoethyl)-glycine units. Because PNA is not recognized by the polymerase as natural DNA, it cannot serve as a primer for polymerization or as a substrate for Taq polymerase exonuclease activity. Since PNA contains no charged phosphate groups, the binding between PNA and DNA is stronger than between DNA and DNA due to the lack of electrostatic repulsion. Owing to the uncharged PNA backbone, the melting temperature of a perfectly matched PNA–DNA duplex displays higher than that of DNA–DNA of the same length, although a single mismatch destabilizes the PNA–DNA hybrids, particularly at low salt concentrations, causing a melting temperature shift of 10–18 °C [Citation16,Citation17]. Based on these features, through a PCR clamping approach, primers can be replaced by PNA hybridization probe that is specific for the wild type; PNA probe can suppress the amplification of wild-type DNA, allowing for increased amplification of the mutant allele. KRAS mutations have been described mainly in seven types mutant at exon 2 (codons 12 and 13) and in rare cases at exon 3 (codon 61). In this study, the objective is to develop and evaluate a highly sensitive and simple method using real-time PNA-mediated PCR clamping assay, which enables the accurate and rapid detection of all KRAS codon 12 and 13 mutations in a single reaction.
Materials and methods
Templates and reagents
In this study, each of the seven mutations (shown in ) was engineered into a clone of the wild-type KRAS gene by PCR-based site-directed mutagenesis. These mutants were then cloned into the plasmid pGEM-T (Promega, Tokyo, Japan), for use as template DNA in PNA-mediated PCR clamping assay development. The sequences of the synthesized mutant templates were verified by direct sequencing. These templates were suspended in ddH2O and stored at −20 °C before including in the assays. Double-distilled water (≤18.2 MΩ) was used in all solutions with a Milli-Q system (Millipore, Bedford, MA, USA), unless otherwise stated. Oligonucleotides were synthesized by Purigo Biotech (Taipei, Taiwan) and were further purified by high-performance liquid chromatography (HPLC).
Table 1. Seven types of KRAS mutations used in this study.
PNA probe synthesis
The PNA probes were synthesized by the Panagene (Panagene Inc., Daejeon, Korea) technology, using benzothiazole-2-sulfonyl as the amine-protecting group. The purity of 99.9% PNA probes was ensured by HPLC (Agilent 1100; Agilent Technologies, Wilmington, DE, USA), the quality of which was assessed by MALDITOF (AX1MA-CFR; Shimadzu Co, Kyoto, Japan). The concentration of the PNA probe was determined using a Nano-Drop ND-1000 spectrophotometer (Nano Drop Technologies, Wilmington, DE, USA) [Citation18].
PNA-mediated real-time PCR clamping assay
The PNA-mediated PCR clamping assay was performed in a CFX96 apparatus (Bio-Rad Laboratories, Hercules, CA, USA) according to the manufacturer's instructions. Briefly, 10 mmol/L Tris-HCl (pH8.3), 50 mmol/L KCl, 3.75 MgCl2, 150 µmol/L dNTP, 1 µmol/L of primer forward (5'-TATTATAAGGCCTGCTG-3') and 1 µmol/L primer reverse (5'-TTGGATCATATTCGTCC-3'), 0.75 µL of 1:200 SYBR Green I in dimethyl sulfoxide, 0.7 µmol/L PNA (5’), 1 U of Q5@ high-fidelity DNA polymerase (New England Biolabs, Beverly, MA, USA) and 1 µL of template DNA were mixed in a final volume of 25 µL. The PCR cycling commenced with a 5-min hold at 94 °C followed by 40 cycles of 94 °C for 30 sec, 72 °C for 20 sec and 61 °C for 30 sec. Seven KRAS mutation types were detected using PNA-mediated real-time PCR clamping in this study. The efficiency of PNA-mediated PCR clamping was determined by measuring the threshold cycle (Ct) value. The Ct values for the control and mutation assays were obtained by observing the SYBR Green amplification plots. The delta Ct (ΔCt) value was calculated ([Control Ct] − [Sample Ct] = ΔCt), ensuring that the sample and control Ct values were from the sample to analyse and wild-type control sample. The cut-off ΔCt was defined as 2 for the seven mutation types.
Results and discussion
Establishment of the PNA-mediated real-time PCR clamping assay
PNA-mediated real-time PCR clamping relies on the unique properties of PNA probes. When a mutation in target gene is present, the DNA–PNA duplex is destabilized, allowing strand elongation from a bound DNA oligomer. Therefore, the positive reaction in real-time PCR results from the samples harbouring mutant allele, while the wild-type allele is suppressed [Citation15]. In this method, the reaction signals were observed by intercalation of SYBR Green fluorescent dye. This approach is schematically presented in . The PNA probe was used to suppress amplification of the KRAS wild-type allele ((A)), thereby enhancing preferential amplification of mutant sequences by competitively inhibiting the primer binding to wild-type allele. The PCR efficiency was determined by measuring the Ct value, which was obtained by observing the SYBR Green amplification plots ((B)). The mutant allele was smoothly amplified with the low Ct (24.01), contrast to the wild-type allele suppressed by PNA inhibition with the high Ct (35.51).
Figure 1. The peptide nucleic acid (PNA)-mediated real-time PCR clamping assay. (A) The scheme of the PNA-mediated real-time PCR clamping assay. PNA inhibits wild-type by hybridizing normal sequences, and mutant allele is preferentially amplified. (B) The signal was obtained by observing the SYBR Green amplification plots. RFU, relative fluorescence units.
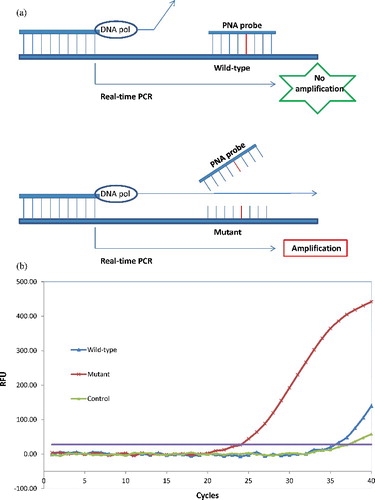
The PNA probe was designed to hybridize fully to the wild-type KRAS allele, referred in the publication [Citation19]. PNA probe hybridization securely inhibits the amplification of the wild-type KRAS allele, while the PNA/mutant-type allele hybrid is unstable, as a result of base pair mismatch, and therefore, does not inhibit Taq polymerase from the extension [Citation20]. Therefore, PNA cannot inhibit completely the wild-type allele amplification under the normal PCR different temperatures cycles. However, the changes in Ct values (ΔCt), calculated as the Ct values of the samples minus the Ct value of the PNA control, could be used to estimate the mutant probability of the analysed sample. The high ΔCt value means that the mutant was efficiently amplified. In this study, the cut-off value of 2.0 was used for determining the presence of mutant DNA.
Optimization of the PNA-mediated real-time PCR clamping assay
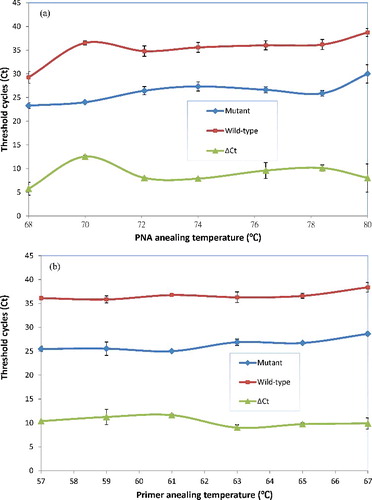
For optimization of the PNA annealing to the full-match hybridized with wild-type template, the normal real-time PCR cycle was expanded with a PNA annealing step between the template denaturation and the primer annealing. During this PNA annealing step, it only allows the PNA to hybridize with its target template at a higher temperature than the step of PCR primer annealing [Citation18]. To optimize the PNA annealing and primer annealing steps for this PNA-mediated real-time PCR clamping, the different PNA annealing temperatures (68–80 °C) and primer annealing temperatures (57–65 °C) on Ct and ΔCt (control Ct–mutant Ct) were investigated, respectively.
As shown in , there was an optimal 70 °C for PNA annealing temperature, which achieved the largest space between the Ct value (24.01 ± 0.16) in mutant-type template and the Ct value (36.52 ± 0.42) in wild-type template ((A)); there was an optimal 61 °C primer annealing temperature, the Ct value (25.02 ± 0.17) in mutant-type template and the Ct value (36.76 ± 0.24) in wild-type template, which achieved the best compromise of Ct value ((B)). From the above results, the optimal annealing temperatures of PNA and primer were 70 and 61 °C, respectively. These PCR conditions were used later for evaluation and detection of KRAS mutations by PNA-mediated real-time PCR clamping assay.
Figure 2. Optimization of the annealing temperatures of peptide nucleic acid (PNA) and primer. (A) Optimization of PNA annealing temperatures for efficient PNA-mediated real-time PCR clamping. (B) Optimization of primer annealing temperatures for efficient PNA-mediated real-time PCR clamping.
Detection of seven mutant alleles by the PNA-mediated PCR clamping assay
The PNA-mediated real-time PCR clamping assay was engineered to detect all seven known mutant alleles of KRAS codons 12 and 13. This design assumption was examined using the seven mutant templates made by PCR-based site-directed mutagenesis. As shown in , the results demonstrated approximately equal efficiencies for the amplification of all seven mutant alleles of KRAS codons 12 and 13. The assay showed little to no bias, validating its effectiveness as a general screening tool for the seven mutations of interest.
Figure 3. KRAS codons 12 and 13 mutant alleles detection of seven known variants by the peptide nucleic acid (PNA)-mediated real-time PCR clamping assay. Amplification of seven mutant sequences corresponding to known mutations of KRAS codons 12 and 13 is shown.
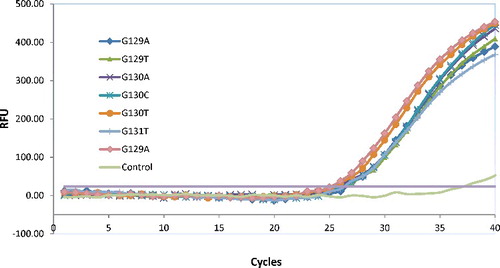
The KRAS mutations have been reported to be mainly seven alleles at codons 12 and 13 [Citation21], which by the artificial refractory mutation system (ARMS)–PCR design approaches using discrimination primers and probes with seven separate assays [Citation22]. For routine clinical benefit, in this study, the PNA competition probe was used to suppress the amplification of the wild-type allele, which made it possible to amplify all seven mutant alleles equally with a single primer set in a reaction tube [Citation17]. This approach greatly reduced the screening cost of the clinical samples, showing obvious superiorities in clinical use.
Sensitivity and specificity of the PNA-mediated real-time PCR clamping assay
For evaluation of the specificity of this assay, various amounts of a wild-type KRAS allele were mixed with a different mutant plasmid, containing each of the seven KRAS mutant alleles. From the results, there were no false-positive or false-negative results observed (data not shown). As the benefit of approximately equal efficiencies for the detection of all seven mutant alleles of KRAS codons 12 and 13, one mutant KRAS allele (G129T) was chosen to evaluate the assay sensitivity using different dilutions of the plasmid containing G129T KRAS mutant allele. The results of the PNA-mediated real-time PCR clamping assay used to detect KRAS mutant DNA diluted by the wild-type DNA were shown in
Figure 4. Sensitivity of the peptide nucleic acid (PNA)-mediated real-time PCR clamping assay for detection of KRAS mutations at codon 12 and 13.
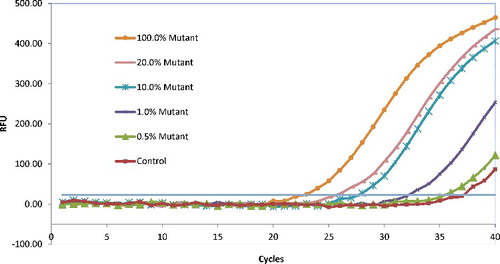
The proportions of mutant DNA to normal DNA were adjusted to each of 100%, 20%, 10%, 1%, 0.5% and 0% (control), and in total 50-pg DNA samples of genomic DNA were used as a template for PNA clamping. As shown in , the ΔCt were 14.47, 11.37, 9.52, 3.97 and 1.70, corresponding to the mutant percentage of 100%, 20%, 10%, 1% and 0.5% in the total templates, respectively; it meant that this assay could detect mutations in the presence of up to 1% of the mutant allele in a mixture of the wild-type KRAS allele.
It is reported that the sensitivity of direct sequencing depends on the percentage of tumour cells in the analysed sample, generally up to 50% tumour cells requested with a detection threshold of 25% mutant DNA into a wild-type environment [Citation21,Citation23,Citation24]. The HRM analysis has been recently reported for the KRAS mutations detection, it can be compromised by a low proportion of tumour cells in the analysed sample and by the difficulty to detect homozygous mutations, but the sensitivity of detection was only 5%–10% [Citation25]. In this study, the sensitivity of the PNA-mediated real-time PCR clamping assay is as low as 1% of a mutant allele in a mixture of mutant- and wild-type DNA, and might correspond to a more sensitive test, applicable for routine purposes without macro- or micro-dissection. The amount of DNA template required was as low as 0.5–50 pg per reaction.
Compared to the other methods, the first significant advantage of this approach is the rapid turnaround time for amplification and detection. The total assay time is short as it requires only 1.5 hours after the sample preparation. The second advantage is its simplicity and cost-effectiveness. Our approach employed the double-strand DNA-binding dye (SYBR Green I) for real-time detection of the PCR reaction process and product, whereas other systems use fluorogenic probes. Since KRAS mutation detection is a fundamentally qualitative detection, it is believed that the simplicity, flexibility and cost-effectiveness of detection using SYBR-Green I may be sufficient for the purpose. Besides their relative expense, the use of fluorogenic probes complicates modification and optimization of real-time PCR, and primers/probes need to be designed according to specific rules, packaged by manufacturers, and are often available through outside reference laboratories at relatively expensive rates [Citation26]. The turnaround time for receiving results is near one week, which can sometimes make a bottleneck for immediately starting anti-EGFR therapy in patients. Moreover, the cost of the testing renders repeated examination impossible; yet, this may sometimes be required for patients in whom the disease recurs after prior anti-EGFR therapy [Citation27]. Repeated testing for KRAS mutations is in demand, particularly for recurrent or metastatic lesions, to allow selection of optimal treatment [Citation28,Citation29]. Meanwhile, this approach can detect seven mutant alleles equally with a single primer set in a reaction tube, greatly cut down the cost of the clinical samples screening by seven separate assays with discrimination primers and probes [Citation30]. Since this approach is simple and less expensive, it may enable clinicians to request repeat tests for KRAS mutations for a given patient. The third significant advantage of this approach is highly sensitive, especially benefiting in the detection of low-level mutant allele in the presence of high level of wild-type environment. Based on these features, this assay could also be proposed to detect the other known mutations and the small amount of mutant alleles which are not able to detect by direct sequencing, and provide clinical information which contributes to therapeutic decisions for patients.
Conclusions
The present PNA-mediated real-time PCR clamping assay is a simple, cost-effective and sensitive method for rapid detection of KRAS mutations. The high sensitivity could be helpful for specimens of CRC without removing contaminated normal cells, which could be proposed for identifying metastatic CRC patients in whom metastases will respond to EGFR-targeted monoclonal antibody therapy.
Disclosure statement
The authors indicated no potential conflicts of interests.
Additional information
Funding
References
- Jemal A , Bray F , Center MM , et al. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90.
- Tan C , Du X . KRAS mutation testing in metastatic colorectal cancer. World J Gastroenterol. 2012;18(37):5171–5180.
- Siena S , Sartore-Bianchi A , Di Nicolantonio F , et al. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J Natl Cancer Inst. 2009;101(19):1308–1324.
- Massarelli E , Varella-Garcia M , Tang X , et al. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clin Cancer Res. 2007;13(10):2890–2896.
- Hoshi K , Takakura H , Mitani Y , et al. Rapid detection of epidermal growth factor receptor mutations in lung cancer by the SMart-Amplification process. Clin Cancer Res. 2007;13(17):4974–4983.
- Miyano S , Hanazawa K , Kitabatake T , et al. Detecting KRAS mutations in peripheral blood of colorectal cancer patients by peptide nucleic acid clamp PCR. Exp Ther Med. 2012;4(5):790–794.
- Krypuy M , Newnham GM , Thomas DM , et al. High resolution melting analysis for the rapid and sensitive detection of mutations in clinical samples: KRAS codon 12 and 13 mutations in non-small cell lung cancer. BMC Cancer. 2006;6:295.
- Guha M , Castellanos-Rizaldos E , Liu PF , et al. Differential strand separation at critical temperature: a minimally disruptive enrichment method for low-abundance unknown DNA mutations. Nucleic Acids Res. 2013;41(3):e50.
- Zhao X , Li Y , Park M , et al. Loop-mediated isothermal amplification assay targeting the femA gene for rapid detection of Staphylococcus aureus from clinical and food samples. J Microbiol Biotechnol. 2013;23(2):246–250.
- Zhao X , Yu CS , Tsai WH , et al. Improvement of the sensitivity of the surface plasmon resonance sensors based on multi-layer modulation techniques. Opt Commun. 2015;335:32–36.
- Zhao X , Tsao Y , Lee F , et al. Optical fiber sensor based on surface plasmon resonance for rapid detection of avian influenza virus subtype H6: initial studies. J Virol Methods. 2016;233:15–22.
- Milbury CA , Li J , Makrigiorgos GM . PCR-based methods for the enrichment of minority alleles and mutations. Clin Chem. 2009;55(4):632–640.
- Zhao X , Lin CW , Wang J , et al. Advances in rapid detection methods for foodborne pathogens. J Microbiol Biotechnol. 2014;24(3):297–312.
- Zhao X , Wei C , Zhong J , et al. Research advance in rapid detection of foodborne Staphylococcus aureus . Biotechnol Biotec EQ. 2016; DOI: 10.1080/13102818.2016.1209433.
- Demers DB , Curry ET , Egholm M , et al. Enhanced PCR amplification of VNTR locus D1S80 using peptide nucleic-acid (PNA). Nucleic Acids Res. 1995;23(15):3050–3055.
- Kyger EM , Krevolin MD , Powell MJ . Detection of the hereditary hemochromatosis gene mutation by real-time fluorescence polymerase chain reaction and peptide nucleic acid clamping. Anal Biochem. 1998;260(2):142–148.
- Tatsumi K , Mitani Y , Watanabe J , et al. Rapid screening assay for KRAS mutations by the modified smart amplification process. J Mol Diagn. 2008;10(6):520–526.
- Choi JJ , Cho M , Oh M , et al. PNA-mediated real-time PCR clamping for detection of EGFR mutations. B Korean Chem Soc. 2010;31(12):3525–3529.
- Dabritz J , Hanfler J , Preston R , et al. Detection of Ki- ras mutations in tissue and plasma samples of patients with pancreatic cancer using PNA-mediated PCR clamping and hybridisation probes. Br J Cancer. 2005;92(2):405–412.
- Jeong D , Jeong Y , Park JH , et al. BRAFV600E mutation analysis in papillary thyroid carcinomas by peptide nucleic acid clamp real-time PCR. Ann Surg Oncol. 2013;20(3):759–766.
- Beau-Faller M , Legrain M , Voegeli AC , et al. Detection of K- Ras mutations in tumour samples of patients with non-small cell lung cancer using PNA-mediated PCR clamping. Br J Cancer. 2009;100(6):985–992.
- Domagala P , Hybiak J , Sulzyc-Bielicka V , et al. KRAS mutation testing in colorectal cancer as an example of the pathologist's role in personalized targeted therapy: a practical approach. Pol J Pathol. 2012;63(3):145–164.
- Pao W , Laclanyi M . Epidermal growth factor receptor mutation testing in lung cancer: searching for the ideal method. Clin Cancer Res. 2007;13(17):4954–4955.
- Eberhard DA , Giaccone G , Johnson BE . Biomarkers of response to epidermal growth factor receptor inhibitors in non-small-cell lung cancer working group: standardization for use in the clinical trial setting. J Clin Oncol. 2008;26(6):983–994.
- Do H , Krypuy M , Mitchell PL , et al. High resolution melting analysis for rapid and sensitive EGFR and KRAS mutation detection in formalin fixed paraffin embedded biopsies. Bmc Cancer. 2008;8:142.
- Nitsche A . Oligonucleotide design for in-house real-time PCR applications in microbiology. In: Mackay IM , editor. Real-time PCR in microbiology. Norfolk (VA): Caister Academic Press; 2007. p. 41–69.
- Takata M , Chikumi H , Matsunami K , et al. A new rapid method for detecting epidermal growth factor receptor mutations in non-small cell lung cancer. Oncol Rep. 2015;33(3):1040–1048.
- Sharma A , Zhang G , Aslam S , et al. Novel approach for clinical validation of the cobas KRAS mutation test in advanced colorectal cancer. Mol Diagn Ther. 2016; 20(3):231–240.
- Dudley J , Tseng LH , Rooper L , et al. Challenges posed to pathologists in the detection of KRAS mutations in colorectal cancers. Arch Pathol Lab Med. 2015;139(2):211–218.
- Qu S , Liu L , Gan S , et al. Detection of low-level DNA mutation by ARMS-blocker-Tm PCR. Clin Biochem. 2015;49(3):287–291.