
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Culture-dependent and culture-independent methods were compared and evaluated in the study of the endophytic diversity of Dendrobium officinale. Culture-independent methods consisted of polymerase chain reaction–denaturing gradient gel electrophoresis (PCR-DGGE) and metagenome methods. According to the results, differences were found between the three methods. Three phyla, namely Firmicutes, Proteobacteria, and Actinobacteria, were detected using the culture-dependent method, and two phyla, Firmicutes and Proteobacteria, were detected by the DGGE method. Using the metagenome method, four major phyla were determined, including Proteobacteria (76.54%), Actinobacteria (18.56%), Firmicutes (2.27%), and Bacteroidetes (1.56%). A distinct trend was obtained at the genus level in terms of the method and the corresponding number of genera determined. There were 449 genera and 16 genera obtained from the metagenome and DGGE methods, respectively, and only 7 genera were obtained through the culture-dependent method. By comparison, all the genera from the culture-dependent and DGGE methods were contained in the members determined using the metagenome method. Overall, culture-dependent methods are limited to ‘finding’ endophytic bacteria in plants. DGGE is an alternative to investigating primary diversity patterns; however, the metagenome method is still the best choice for determining the endophytic profile in plants. It is essential to use multiphasic approaches to study cultured and uncultured microbes.
Introduction
Dendrobium officinale Kimura et Migo, of the family Orchidaceae, which is mainly distributed in South-east and South Asian countries, is a traditional Chinese medicinal plant associated with immunostimulating [Citation1] and anti-tumour [Citation2] functions. D. officinale usually grows on nutrient-poor cliffs on mountains at altitudes between 900 m and 1500 m, mainly in the Yunnan, Sichuan, Guizhou, Guangxi, and Zhejiang provinces of China [Citation3]. In the wild, it grows slowly, and microbes may provide vital nutrition for it as they do for other orchids.
Fungi were reported to play a critical role in the growth of D. officinale [Citation4–Citation6]. In addition, prokaryotes are directly related to the function of plants, and varied endophytes were found in D. officinale. Therefore, clarification of the diversity and function of the endophytes may more effectively help to elucidate their roles in their hosts.
Culture-dependent methods are traditionally used to obtain bacteria and thus to study their roles. However, less than 1% of bacteria can be usually be cultured, which is detrimental to the study of the comprehensive roles of endophytic bacteria. More recently, rapid developments in molecular microbial ecology have allowed traditional microbial research approaches to be complemented by the collection of unprecedented quantities of 16S rRNA data [Citation7]. Molecular methods, such as sequencing of SSU ribosomal DNA, single-strand conformation polymorphism (SSCP), amplified ribosomal DNA restriction analysis (ARDRA), random amplified polymorphic DNA (RAPD), terminal-restriction fragment length polymorphism (T-RFLP), denaturing and temperature gradient gel electrophoresis (DGGE/TGGE), and novel bar-coded amplicon pyrosequencing (metagenome) [Citation8] are becoming increasingly popular in the studies of communities of microorganisms [Citation9,Citation10]. All these methods are usually based on the 16S rRNA gene, and they are generally suitable for the analysis of environmental samples.
The present paper compares three popular methods, namely the culture-dependent method, DGGE, and metagenome sequencing. The purpose of this study is to evaluate these methods and thus provide solid guidance for the correct selection of the proper methods for the study of microbial communities. Of course, the selection of the proper method also leads to better modification of the appropriate culturing method for obtaining much more bacteria, which is vital for the discovery of functional bacteria and their corresponding uses.
Materials and methods
Sampling
The root, stem, and leaf samples of D. officinale were collected in September 2013 from a planting base in Wenzhou, Zhejiang Province, China (28°15′ N, 121°1′ E). Samples were obtained from completely randomized locations at the same time using a serpentine sampling method and transported to the laboratory on the same day. All the sampled plants had a characteristically healthy appearance and were used for endophytic bacterial diversity analysis.
The D. officinale plants were washed under running water to remove adhering soil particles and the majority of impurities. Root, stem, and leaf tissues were separated and surface-sterilized by immersion in 70% ethanol for 1 min, followed by 1% sodium hypochlorite for 3 min, and, finally, washed three times in sterile-distilled water. The root, stem, and leaf samples of D. officinale were designated as WZR, WZS, and WZL, respectively.
DNA extraction
Total genomic DNA was extracted from 0.2 g fresh plant tissues using the modified cetyltrimethyl ammonium bromide (CTAB) method [Citation11], with the incubating time at 65 °C prolonged to 90 min. The DNA concentration in each sample was measured using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) to verify that the samples had similar concentrations for amplification. The DNA was stored at −20 °C.
Diversity profiling based on culture-dependent method
Roots, stems, and leaves of the surface-sterilized plants were separately ground with a mortar and pestle and diluted with saline phosphate buffer. A series of dilutions (10−1, 10−2, 10−3, 10−4, and 10−5) were spread on nutrient agar (NA) plates, with 200 μL per plate and three repetitions per dilution. After incubation at 28 °C for 48 h, morphologically different colonies were selected as candidate antagonists.
Diversity profiling based on PCR-DGGE method
Nested-PCR-DGGE was carried out as previously described, with some modification [Citation11]. Briefly, the bacterial-specific 16S rRNA fragment was first amplified using primers fM1 (5’-CCGCGTGNRBGAHGAAGGYYYT-3’) and rC5 (5’-TAATCCTGTTTGCTCCCCAC-3’). Subsequently, the initial PCR products were used as templates for a nested-PCR collectively using 515f (GTGCCAGCMGCCGCGGTAA) and rC5. Therein, primer 515f was linked with a GC clamp (5’-CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGCACGGGGGG -3’) to create a DNA fragment suitable for DGGE analysis.
PCR reactions were performed in a 50 μL reaction mixture, containing 20 pmol of each primer, 10 nmol of each deoxynucleoside triphosphate, 5 μL of 10 × PCR buffer (100 mM Tris-HCl [pH 9.0], 15 mM MgCl2, 500 mmol/L KCl, 1% [vol/vol] Triton X-100, 0.1% [wt/vol] gelatin), 2 U of Taq DNA polymerase (Dongsheng Biotech, China), and 1 μL of template DNA. The reactions were carried out under the following conditions: 94 °C for 2 min, followed by 35 cycles at 94 °C for 30 sec, 57 °C for 30 sec, 72 °C for 30 sec, and, finally, 72 °C for 8 min. Electrophoresis was performed to confirm successful amplification.
The PCR products from the nested PCR were purified using a MiniElute PCR Cleanup Kit HG-1006 (Haogene Biotech (Hangzhou) Co., LTD), and analysed by DGGE using the DCODE Universal Mutation Detection System (Bio-RAD, Hercules, CA, USA) following the manufacturer's instructions. Gels contained 6% acrylamide/bis-acrylamide with a denaturing concentration ranging from 40% to 60%. Electrophoresis was performed in TAE 1 × buffer at 60° C at 100 V for 12 h. Bands were visualized using a silver staining kit and photographed. Bands were purified from the gels and DNA was incubated at 37 °C for 15 min in 50 μL of sterile water and eluted overnight at 4 °C. The DNA was re-amplified with the nested-primer pair 515f/rC5 without the GC clamp. The PCR products were sequenced by the Shanghai Sonny Biological Technology Company. The sequences were analysed using the BLASTN algorithm in the GenBank database.
Diversity profiling based on metagenomic sequencing
The V4 hypervariable region of the bacterial 16S rRNA genes was amplified using primers fM1 and rC5. Electrophoresis was performed to confirm successful amplification, and PCR products were extracted from the agarose gel by a Qiagen QIAquick gel extraction kit (Qiagen, CA). DNA extraction from filters was performed following standard procedures [Citation10]. The PCR products were subjected to AxyPrepTM Mag PCR Normalizer normalization processing and used to build an Illumina sequencing library. By covering the rehabilitate with exonuclease activity to repair the end, a flat end was produced. An ‘A’ base was then added to the end of the flat end, which was then connected with an Illumina sequencing end-matched connector. These joints consisted of two unique sequencing primers with hybridization sites. The additional sequence which complemented the oligonucleotides in the flowing channel was added to the joint sequence which had the PCR primer tail, then selected and purified based on the size of the gel. The completed sequence library was loaded into the cBot or clusters system, using clusters and Miseq sequencing.
Data analysis
PCR products were analysed by computer sequencing of segments reading a length of 100 bp, and the results showed that the sample sequencing error rate was low, which proved that the D. officinale sample DNA bases were of good quality, thus ensuring that the sequencing results were of high accuracy. The raw data sequencing was performed by removing the sequences whose lengths did not meet the minimum quality filter criteria, removing the barcode sequences and the joint sequences, thus acquiring high-quality, credible and clean data. The QC reads were de-noised using the ‘pre.cluster’ routine (http://www.Mothur.org/wiki/Pre. cluster) to remove sequences that were likely due to sequencing errors. Unique sequences were then aligned against the SILVA database and chimeric sequences were removed using uchime. After sequence quality control and filtration, the effective sequences of each sample were clustered using cdhit to build operational taxonomic units (OTUs) with a cut-off value of 97% sequence identity. In order to analyse species composition more accurately, the RDP Classifier system was used for all OTU representative sequences for species classification analysis. Rarefaction curves were plotted using MG-RAST [Citation12]. The Venn diagram was constructed according to the metagenome analysis data. The Shannon (H′), Simpson (D), and Chao1 indices for species richness of community were calculated as follows:
where S is the total number of OTUs, ni is the number of the ith OTU, N is the number of total OTUs in the sample, Sobs is the number of detected OTUs, F12 is the number of OTUs including one sequence, and F2 is the number of OTUs including two sequences.
Results and discussion
Diversity of endophytic bacteria based on the culture-dependent method
Diversity profiles of endophytic bacteria from different tissues of D. officinale were analysed using a culture-dependent method. In total, 17 bacterial isolates were obtained from D. officinale, including 8 isolates from WZR, 5 from WZS, and 4 from the WZL sample (). All the isolates showed 99%–100% similarity with a specific genus or species.
Table 1. The identification and classification of endophytic bacteria based on culture-dependent and DGGE methods.
On the phylum level, differences existed among the different tissue types. As shown in , all of the 17 isolates belonged to three phyla; namely Firmicutes, Proteobacteria, and Actinobacteria. Most of the isolates from WZR belonged to Firmicutes, and some belonged to Proteobacteria. The isolates from WZL were grouped into the above three phyla, while only Actinobacteria was found among the isolates from WZS.
Figure 1. Comparison of phylum level distributions for three tissue types from culture and DGGE methods.
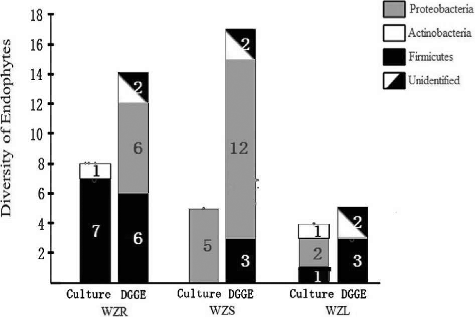
Different bacterial profiles on the genus level were found among the different tissues of D. officinale. The results showed that most of the isolates from WZR belonged to Bacillus or closely related genera, such as Paenibacillus, while the other two isolates belonged to Sporosarcina contaminans and Brevibacterium sp. Interestingly, five isolates from WZS were all identified as Burkholderia. The isolates from WZL were identified as Methylobacterium, Curtobacterium, and Bacillus. Obviously, different genera came from different tissues. The bacterial diversity of WZR was greater than that of the other two tissue types.
Diversity of endophytic bacteria based on DGGE
The corresponding DGGE patterns indicate that the tested samples of D. officinale from Wenzhou have a large number of bands. Fourteen, 17, and 13 bands were obtained from WZR, WZS and WZL, respectively. More bands were obtained from WZS than from WZR, and WZR shared some common bands with WZS and WZL. The diversity analysis was repeated in triplicate and the same pattern was obtained each time. Therefore, the bands were purified for subsequent sequencing and identification.
At the phylum level, the distribution of endophytic bacteria was similar among different tissues. As shown in , the identified endophytes from the three tissues belonged to two phyla; namely Firmicutes and Proteobacteria, as well as a third group of unidentified phyla. Among the 14 fragments from WZR, there were 6 fragments each belonging to Firmicutes and Proteobacteria. However, more fragments belonged to Proteobacteria than to Firmicutes in both WZS and WZL. The other fragments, including two from WZR, two from WZS, and three from WZL, shared less than 30% homology with the known species and were different from each other. Therefore, they were divided into a separate, unknown group.
Among the 44 fragments, 32 fragments were identified as definite species or genera with over 97% nucleotide identity (). The majority of the fragments belonged to Proteobacteria, including Rhizobium, Ochrobactrum, Pseudaminobacter, Klebsiella, Brevundimonas, Burkholderia, Enterobacteriales, Delftia, and Pseudochrobactrum. Pseudaminobacter was the most commonly occurring, with six fragments identified as this genus. The genera Burkholderia (five fragments), Delftia (four fragments), and Pseudochrobactrum (three fragments) followed accordingly in terms of abundance. The identified genera, with the exception of Tissierella, Pseudaminobacter, and Delftia, were unevenly distributed in WZR, WZS, and WZL. Some genera were found only in a specific tissue. For example, Enterococcus, Brevibacillus, Rhizobium, Ochrobactrum, and Klebsiella were restricted to WZR, Burkholderia and Brucella to WZS, and Staphylococcus was restricted to WZL. The other genera were shared by two of the tissues.
Diversity of endophytic bacteria based on metagenome method
After quality control and filtering, Illumina Miseq sequencing analysis produced 392,474 sequences with good quality (124,437 from WZG, 122,918 from WZS, and 145,119 from WZL). The resulting sequences were grouped in 89,526 OTUs (19,460 from WZG, 28,290 from WZS, and 41,776 from WZL). For all three of the sample types, the rarefaction curves nearly reached a plateau, indicating that the sampling depth and sequencing coverage were good (Figure 1S), and the highest species richness of WZG is evident in Figure 1S. Shannon, Simpson, and Chao1 indices also confirmed the highest bacterial diversity for WZG, followed by WZL and WZS (Table 1S).
Diversity of endophytic bacteria on phylum level
Overall, as shown in , the most predominant phylum across all three sample types was Proteobacteria, which accounted for 78.31% of the total phyla. The other three dominant phyla were Firmicutes (12.42%), Actinobacteria (6.48%), and Bacteroidetes (1.80%).
Figure 2. Comparison of phylum level distributions for three tissue types based on the metagenome method.
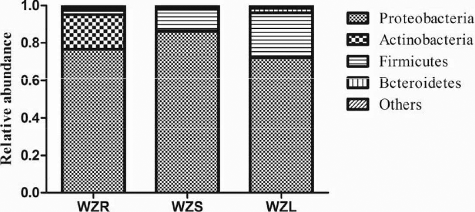
For single tissues, the regularity of the phylum distribution was similar to that of the whole, with some alteration in percentages. In the sample WZR, the dominant phyla were Proteobacteria (76.54%), Actinobacteria (18.56%), Firmicutes (2.27%), Bacteroidetes (1.56%), and the remaining phyla were only 1.17% of the total. For WZS and WZL, the absolute dominant phylum was Proteobacteria, with 86.20% in WZS and 72.18% in WZL, and the second and third most dominant phyla were Firmicutes and Bacteroidetes, with 11.62% and 1.23% in WZS, and 23.36% and 2.62% in WZL, respectively. In the case of Actinobacteria, which was the second most dominant phylum in WZR, the abundance was only 0.31% and 0.58% in WZS and WZL, respectively. In addition, the abundances of Acidobacteria in WZS and WZL were 0.17% and 0.64%, respectively. These results indicate that the bacterial diversity of WZS was more similar to that of WZL than to that of WZR.
Diversity of endophytic bacteria at genus level
In total, there were 449 genera sequenced from the three tissue types of D. officinale, and the average abundance of 23 of the genera was over 1% (). The five dominate genera were Aquamicrobium (10.48%), Brucella (9.62%), Pseudochrobactrum (7.03%), Burkholderia (6.76%), and Acinetobacter (4.34%). For the single tissues, the dominant genera were different from each other. For WZR, there were 20 genera with over 1% abundance, and the 5 dominant genera were Burkholderia (9.34%), Marmoricola (6.30%), Rhodanobacter (6.29%), Methylovirgula (5.82%), and Rhodoblastus (3.92%). There were 15 genera with over 1% abundance, and the dominant genera in WZS were Brucella (18.53%), Aquamicrobium (18.25%), Pseudochrobactrum (11.91%), Burkholderia (10.93%), and Aminobacter (4.23%), which occupied 63.85% of the total genera. For WZL, there were 14 genera with over 1% abundance. The dominant genera were Aquamicrobium (12.77%), Acinetobacter (11.57%), Brucella (10.13%), Pseudochrobactrum (8.83%), Delftia (6.50%), Bacillus (4.76%), Falsibacillus (4.58%), and Brevundimonas (4.07%).
Table 2. Comparison of percentage (%) of the metagenome sequences affiliated with the dominant bacterial genera (average abundance >1%) for three tissue sample types.
Therefore, from the results above, it is evident that different sample types had certain same dominant genera. For example, Burkholderia existed in WZR and WZS, and Brucella, Aquamicrobium, and Pseudochrobactrum, the most dominant genera from Alphaproteobacteria, were found to be abundant in both WZS and WZL. In addition, 36 genera, such as Bacillus, Falsibacillus, Ornithinibacillus, Cerasibacillus, Brevibacillus, and other related genera, were distributed mainly in WZS and WZL, especially in the leaves. On the other hand, different samples had their own special or unique genera. Based on previous reports [Citation13,Citation14], a rare species may be defined as a frequency of species with 0.01% of the total population, and the rest of that population is considered to be abundant. In the present study, there were 115 genera with over 0.01% total population found only in WZR, such as Rhodanobacter (6.29%), Methylovirgula (5.82%), Rhodoblastus (3.92%), Afipia (0.35%), and Methylocapsa (0.97%). For WZS, there were 7 unique genera with over 0.01% abundance, including Salipiger (0.01%), Rheinheimera (0.01%), Novosphingobium (0.01%), Sphingopyxis (0.01%), and Pseudoxanthomonas (0.01%). In the sample WZL, there were 30 unique genera with over 0.01% abundance, such as Alistipes (0.03%), Cedecea (0.02%), Paucisalibacillus (0.02%), Ethanoligenens (0.02%), and Parabacteroides (0.01%).
Evidently, WZR had special endophytic bacteria with more diversity, and the dominant genera shared by WZR and WZS were only Burkholderia and Methyloversatilis. However, WZS and WZL had quite similar structures of bacterial endophytes, e.g. similar types and percentages. They shared 10 genera with over 1% abundance, especially, the same three dominant genera Brucella, Aquamicrobium, and Pseudochrobactrum. This suggests that although all the samples are geographically linked to each other, the composition of the bacterial communities at the genus level varied between them.
Culture-dependent versus DGGE or Illumina sequencing
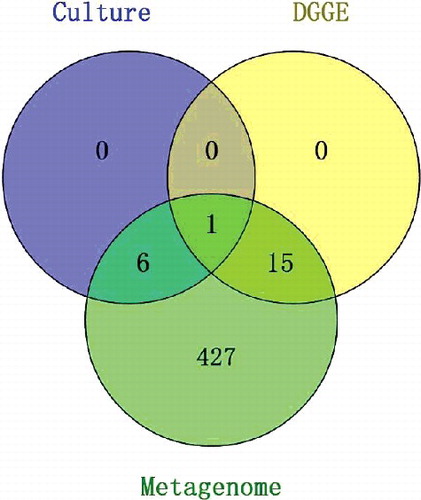
According to the results, differences were found between the three methods, especially on the genus level (). Three, two, and four phyla were detected with the culture-dependent, DGGE, and metagenome methods, respectively. Proteobacteria and Firmicutes were observed to be predominant in all three tissue types of D. officinale, irrespective of the method used. Our findings with regard to complexity and composition are consistent with those of earlier studies of rhizosphere [Citation15,Citation16], paddy soils [Citation17], endophytic bacteria in potato [Citation18], and water samples [Citation19]. Nevertheless, when the most dominant phyla of the bacterial isolates were compared using DGGE and Illumina sequencing, it became clear that different abundances were being targeted by each of the methods. There were 7 and 16 genera identified through culturing and PCR-DGGE, respectively. Based on metagenome results, 449 genera were sequenced from the three tissues of D. officinale, with 23 of the genera having an average abundance of over 1%. All the genera identified from the culture and PCR-DGGE methods overlapped with the genera from the metagenome method. However, 3 of the 7 genera from the culture method and 7 of the 16 genera from PCR-DGGE were included in the 23 dominant genera of the metagenome, respectively. For the culture method, only two genera were included in the PCR-DGGE results. These results demonstrate the higher bacterial diversity coverage of Illumina sequencing compared to the culture-dependent and DGGE methods.
Figure 3. Comparison of genus levels using metagenome, culture-dependent, and DGGE methods.
It is evident that the culture-dependent method tended to produce biased results for microbes. The isolates from D. officinale included Bacillus, Paenibacillus, Sporosarcina, Brevibacterium, Burkholderia, Methylobacterium, and Curtobacterium, which are also contained in the genera isolated from other plants, such as alfalfa [Citation20], clover [Citation21], and pea [Citation22]. Similar to the present results, Moreira [Citation23] demonstrated that a culture-dependent diversity survey presented lower coverage than the other methods. Richard [Citation24] declared that culturable bacteria were probably the largest, most active prokaryotes in a given sample [Citation25]. In fact, the nutritional requirements and growth ratios ultimately determine which bacteria can thrive and predominate under the determinative conditions. Therefore, it appears that the predominance of each representative bacterium did vary with the culture medium. Thus, it is necessary to modify the culture media and eliminate the disturbance or interference of rapidly growing bacteria which allow the growth of more bacteria. On the other hand, it is very difficult to model the exact nutritional environment or niche of plants. Therefore, some bacteria that are rigidly dependent on the plant niche can be easily missed [Citation26], which might be one important reason for failing to culture many bacteria, thus underestimating the microbial diversity [Citation2,Citation9,Citation27].
By comparison, it is easy to detect many more genera using culture-independent methods and thus they are more reliable methods for investigating the microbial profiles of communities. In the early 1990s, Muyzer [Citation28] developed a culture-independent method with the potential to determine microbial flora quickly and economically, that was termed DGGE. This approach has the advantage of directly profiling microbial populations present in specific ecosystems by separating PCR products that originated with universal primers [Citation29]. Heuer and Smalla [Citation30] reported that differences in the relative abundance of community members might affect the detection of certain species due to competition during PCR. Edouard [Citation31] demonstrated that DNA extraction yields could vary significantly, even for closely related strains. Furthermore, previous studies showed that species representing less than 1% of the total community were thought not to be visible in the DGGE profiles of the microbial community [Citation32,Citation33]. These studies provide a good explanation for the observations that much less phyla and genera were obtained from DGGE than from the metagenome method. In comparison to DGGE, the pyrosequencing method has a higher capacity for exploring the bacterial richness and diversity and to detect cultured organisms, with the advantage of being less laborious.
A combination of culture-dependent and culture-independent methods might provide a powerful strategy to investigate microbes. Culture-independent microbial diversity analysis revealed the presence of unculturable bacteria in most environmental samples [Citation32,Citation34]. Half of the known phyla in eubacteria are not represented by any cultured member, indicating the vast phylogenetic diversity of these ‘yet to be cultured’ bacteria. Nevertheless, ‘unculturable’ does not mean ‘can never be cultured’; rather, it signifies that we lack critical information on biology of such bacteria, and this presents both challenges and opportunities [Citation33]. It provided the simple explanation as to why these bacteria do not grow in the laboratory; namely that microbiologists are failing to replicate essential aspects of their environments. It was also shown recently that previously uncultivable microorganisms could be grown in pure culture if provided with the chemical components of their natural environment [Citation22,Citation33,Citation35].
Previously unknown types of bacteria can be recovered by the combination of high-throughput cultivation techniques with efficient high-resolution phylogenetic fingerprinting [Citation36,Citation37], which will help us identify and obtain some novel or difficultly cultivable microbes.
Conclusions
The above results show that in terms of endophyte identification, the traditional culture method has certain limitations: the PCR-DGGE method can give more detailed results, while the metagenome method was able to provide a complete and thorough analysis. In our opinion, it is essential to use multiphasic approaches to thoroughly study both ‘cultured and uncultured microbes’. Future detailed investigations will provide more substantial proof of the utility of this combined approach to the study of microbial communities.
Supplementry_Data.pdf
Download PDF (49.4 KB)Acknowledgments
We are grateful to Dr Greg Duns for his valuable comments on the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Chen X, Guo S. Advances in the research of constituents and pharmacology of Dendrobium. Nat Prod Res Dev. 2000;13:70–75.
- Ward DM, Ferris MJ, Nold SC, et al. A natural view of microbial biodiversity within hot spring cyanobacterial mat communities. Microbiol Mol Biol Rev. 1998;62:1353–1370.
- Shiau YJ, Nalawade SM, Hsia CN, et al. In vitro propagation of the Chinese medicinal plant, Dendrobium candidum Wall. Ex Lindl., from axenic nodal segments. In Vitro Cell Dev Biol Plant. 2005;41:666–670.
- Zhang M, Xia H, Zhu L, et al. Research progress of Dendrobium tissue culture. China J Chin Mater Med. 2000;6:323–326.
- Chen L, Zhang QY, Jia M, et al. Endophytic fungi with antitumor activities: Their occurrence and anticancer compounds. Crit Rev Microbiol. 2014;8:1–20.
- Tan XM, Wang CL, Chen XM, et al. In vitro seed germination and seedling growth of an endangered epiphytic orchid, Dendrobium officinale, endemic to China using mycorrhizal fungi (Tulasnella sp.). Sci Hortic. 2014;165:62–68.
- Caporaso JG, Lauber CL, Walters WA, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci. 2011;108:4516–4522.
- Oja J, Kohout P, Tedersoo L, et al. Temporal patterns of orchid mycorrhizal fungi inmeadows and forests as revealed by 454 pyrosequencing. New Phytol. 2015;205:1608–1618.
- Tringe SG, Hugenholtz P. A renaissance for the pioneering 16S rRNA gene. Curr Opin Microbiol. 2008;11:442–446.
- Venter JC, Remington K, Heidelberg JF, et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science. 2004;304:66–74.
- Yu J, Zhou XF, Yang SJ, et al. Design and application of specific 16S rDNA-targeted primers for assessing endophytic diversity in Dendrobium officinale using nested PCR-DGGE. Appl Microbiol Biotechnol. 2013;97:9825–9836.
- Meyer F, Paarmann D, D'Souza M, et al. The metagenomics RAST server – a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386.
- Galand PE, Casamayor EO, Kirchman DL, et al. Ecology of the rare microbial biosphere of the Arctic Ocean. Proc Natl Acad Sci. 2009;106:22427–22432.
- Kaeberlein T, Lewis K, Epstein SS. Isolating "uncultivable" microorganisms in pure culture in a simulated natural environment. Science. 2002;296:1127–1129.
- Kim MC, Ahn JH, Shin HC, et al. Molecular analysis of bacterial community structures in paddy soils for environmental risk assessment with two varieties of genetically modified rice, Iksan 483 and Milyang 204. J Microbiol Biotechnol. 2008;18:207–218.
- Lu Y, Rosencrantz D, Liesack W, et al. Structure and activity of bacterial community inhabiting rice roots and the rhizosphere. Environ Microbiol. 2006;8:1351–1360.
- Shrestha PM, Kube M, Reinhardt R, et al. Transcriptional activity of paddy soil bacterial communities. Environ Microbiol. 2009;11:960–970.
- Garbeva P, Van Overbeek LS, Van V, et al. Analysis of endophytic bacterial communities of potato by plating and denaturing gradient gel electrophoresis (DGGE) of 16S rDNA based PCR fragments. Microb Ecol. 2001;41:369–383.
- Vaz MI, Egas C, Nunes OC, et al. Culture-dependent and culture-independent diversity surveys target different bacteria: a case study in a freshwater sample. Antonie van Leeuwenhoek. 2011;100:245–257.
- Gagné S, Richard C, Rousseau H, et al. Xylem-residing bacteria in alfalfa roots. Can J Microbiol. 1987;33:996–1000.
- Sturz A, Christie B, Matheson B, et al. Biodiversity of endophytic bacteria which colonize red clover nodules, roots, stems and foliage and their influence on host growth. Biol Fert Soils. 1997;25:13–19.
- Elvira RM, Van VJ. Natural incidence of endophytic bacteria in pea cultivars under field conditions. Can J Microbiol. 2000;46:1036–1041.
- Ellis RJ, Morgan P, Weightman AJ, et al. Cultivation-dependent and-independent approaches for determining bacterial diversity in heavy-metal-contaminated soil. Appl Environ Microbiol. 2003;69:3223–3230.
- Bakken LR, Van EJ, Trevors J. Culturable and nonculturable bacteria in soil. In: Elsas JDV, Trevors JT, Wellington EMH, editors. Modern soil microbiology. New York (NY): Marcel Dekker; 1997. p. 47–61.
- Amann RI, Ludwig W, Schleifer KH. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev. 1995;59:143–169.
- Hugenholtz P, Goebel BM, Pace NR. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol. 1998;180:6793–6793.
- Rantsiou K, Urso R, Iacumin L, et al. Culture-dependent and-independent methods to investigate the microbial ecology of Italian fermented sausages. Appl Environ Microbiol. 2005;71:1977–1986.
- Muyzer G, Smalla K. Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie Van Leeuwenhoek. 1998;73:127–141.
- Miambi E, Guyot JP, Ampe F. Identification, isolation and quantification of representative bacteria from fermented cassava dough using an integrated approach of culture-dependent and culture-independent methods. Int J Food Microbiol. 2003;82:111–120.
- Schloss PD, Handelsman J. Status of the microbial census. Microbiol Mol Biol Rev. 2004;68:686–691.
- Stewart EJ. Growing unculturable bacteria. J Bacteriol. 2012;194:4151–4160.
- Connon SA, Giovannoni SJ. High-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Appl Environ Microbiol. 2002;68:3878–3885.
- Rappé MS, Connon SA, Vergin KL, et al. Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature. 2002;418:630–633.
- Quesada ME, Lopez -Diaz C, Landa BB. The hidden habit of the entomopathogenic fungus Beauveria bassiana: first demonstration of vertical plant transmission. PLoS One. 2014;9(2):e89278. http://dx.doi.org/10.1371/journal.pone.0089278
- Kaeberlein T, Lewis K, Epstein SS. Isolating "uncultivable" microorganisms in pure culture in a simulated natural environment. Science. 2002;296:1127–1129.
- Zi XM, Sheng CL, Goodale UM, et al. In situ seed baiting to isolate germination-enhancing fungi for an epiphytic orchid, Dendrobium aphyllum (Orchidaceae). Mycorrhiza. 2014;24(7):487–499.
- Na-monrug K, Kingsley D, Kongkanda C, et al. Using in situ seed baiting technique to isolate and identify endophytic and mycorrhizal fungi from seeds of a threatened epiphytic orchid, Dendrobium friedericksianum Rchb.f. (Orchidaceae). Agric Nat Resour. 2016;1e6: 8–13.