
ABSTRACT
Mutations in KCNQ2 are associated with a range of electroclinical syndromes with dominant inheritance that are differentiated by the age at onset of the seizures and are associated with good prognosis. These are benign familial neonatal seizures (BFNS), benign familial neonatal--infantile seizures (BFNIS) and benign familial infantile seizures. Herein, we report the results of a systematic screening of KCNQ2 in 27 unrelated Bulgarian patients with compatible clinical diagnoses. Two pathogenic point mutations were identified: a novel splice-site c.1526-2A>G variation causing BFNS and a missense c.998G>A alteration in a patient with BFNIS, who subsequently developed benign epilepsy with centro-temporal spikes. Additionally, multiplex ligation-dependent probe amplification analysis and array comparative genomic hybridization assay detected a de novo deletion on 20q13.3 encompassing 0.41 Mb genomic region and covering 11 genes, including KCNQ2 and CHRNA4. This large-scale rearrangement was found in a patient with typical BFNS and no additional developmental abnormalities. Overall, KCNQ2 genetic defects were found in 11% of the patients in our cohort. These findings enrich the genetic epidemiology and mutation spectrum of KCNQ2 and allow adequate genetic counselling in the affected families.
Introduction
Benign familial neonatal seizures (BFNS; OMIM (Online Mendelian Inheritance in Man): 121200), benign familial neonatal--infantile seizures (BFNIS; OMIM: 607745) and benign familial infantile seizures (BFIS) are autosomal dominant epileptic disorders with high penetrance. All three electroclinical syndromes show similar clinical features and tend to remit spontaneously later in life. They are differentiated by the age of onset and the associated gene defect. All affected individuals exhibit normal outcome and good response to antiepileptic drugs.
BFNS is caused by mutations in KCNQ2 and KCNQ3, which encode voltage-gated potassium channels expressed in the brain. To date, more than 80 KCNQ2 alterations are described in BFNS patients, while the mutation spectrum of KCNQ3 is narrower Citation[1].
Benign epilepsy with centro-temporal (Rolandic) spikes (BECTS) is another epileptic syndrome associated with good prognosis Citation[2]. It consists of brief, hemifacial seizures that tend to become generalized when occurring nocturnally. Cases of BFNS with detected mutations in KCNQ2 might subsequently develop BECTS Citation[3–5].
Herein, we report the results of the first systematic screening of KCNQ2 in Bulgarian patients and discuss the molecular defects found, as well as genotype–phenotype correlations in comparison to previously reported individuals for the particular group of epileptic disorders.
Subjects and methods
Case study
The study included 27, both sporadic and family, cases with unprovoked epileptic seizures within the first year of life, which were recruited from the major tertiary hospitals in Bulgaria. Among them, 12 patients were diagnosed with BFNS, 3 with BFNIS, 10 with BFIS and 2 with BECTS. All participants or their legal guardians gave written informed consent. The study was approved by the Ethics Committee of the Medical University of Sofia. The cases with identified mutations are described as follows.
Case 1: A one-year-old boy born from a second normal pregnancy via vaginal delivery. At two days of age, he developed a single clonic seizure lasting several seconds. The second convulsion with the same characteristics was observed the next day accompanied with cyanosis. At the day of hospitalization, three generalized tonic--clonic seizures were registered. Oral phenobarbital treatment was conducted and the seizures subsequently disappeared.
Case 2: A seven-year-old boy born prematurely in the 36th week of gestation from a pathological pregnancy with recurrent genital herpes. At the age of four months, he developed partial tonic--clonic seizures accompanied with eye deviation and cyanosis. Because of the later seizure onset, the diagnosis of BFNIS was assumed. The seizure attacks lasted seven days and progressively became more frequent with duration ranging from several seconds to one minute. Valproic acid, oxcarbazepine and diazepam were implemented with no effect. At the age of 1 year and 10 months, a single convulsion with cyanosis appeared after a head stroke. No seizure attacks were observed before the age of three when, during sleep, he suddenly made moaning sounds with hands clenched into fists. The electroencephalogram showed sharp waves in the left side and the centro-temporal areas. Both latter seizures were with rolandic characteristics. The patient was treated with carbamazepine, which resulted in disappearance of the seizures.
Case 3: A one-year-old girl born after a normal pregnancy. At five days of age, she developed one focal tonic--clonic seizure with cyanosis, eye and head deviation lasting one minute. In the next 24 hours, convulsions with the same characteristics repeated 10 times. They gradually disappeared after phenobarbital treatment. The psychomotor development was normal.
The control group included 100 individuals (60 females and 40 males) between 32 and 85 years of age without any history of epilepsy seizures.
Methods
DNA was extracted from peripheral blood, and sequence analysis of all 17 exons and exon–intron boundaries of KCNQ2 was performed using polymerase chain reaction and Sanger sequencing using ABI 3130xl (Applied Biosystems, Foster City, CA, USA). The sequences were analysed using ABI Sequencing Analysis v5.3 and compared with reference sequence NG_009004.1. To investigate the effect of the novel splice-site mutation, NetGene2 software was used, which is based on neuronal networks prediction algorithms (http://www.cbs.dtu.dk/services/NetGene2/). Furthermore, the 100 control individuals were also screened for the presence of the novel mutation. Multiplex ligation-dependent probe amplification (MLPA) analysis was performed using SALSA MLPA kit P166-C1 (MRC Holland, Amsterdam, The Netherlands). The amplification products were separated by capillary electrophoresis using ABI 3130xl (Applied Biosystems) and evaluated using Coffalyser.NET (MRC Holland). Deletions encompassing the KCNQ2 gene were confirmed by array comparative genomic hybridization (aCGH) using a SurePrint G3 Human CGH Microarray Kit, 4 × 180K (Agilent Technologies, Santa Clara, CA, USA). The visualization of the deleted region and its gene content was performed using the Database of Genomic Variants browser (http://dgv.tcag.ca/dgv/app). Segregation analysis was conducted in all families where potentially pathogenic variants had been found.
Ethics approval
All procedures performed in this study involving human participants were in accordance with the ethical standards of the Medical University of Sofia and/or the National Research Committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants and/or their legal guardians included in this study.
Results and discussion
This is the first extensive investigation of the KCNQ2 gene performed in Bulgaria. We identified three individuals with mutations affecting the KCNQ2 gene among 27 patients with a similar electroclinical phenotype. The estimated frequency of 11% shows common involvement of KCNQ2 mutations in families with neonatal and neonatal--infantile onset of seizures. We did not find any mutations among the BFIS cases, which is consistent with previous studies about the smaller contribution of KCNQ2 mutations to the development of BFIS Citation[6].
Direct sequencing of the KCNQ2 gene revealed two heterozygous pathogenic variants: a novel splice-site mutation c.1526-2A>G in patient 1 ((a)) and a missense mutation р.Arg333Gln (c.998G>A) in patient 2 ((c)).
Figure 1. (a) Electropherogram of family 1 with detected heterozygous splice-site mutation c.1526-2A>G, located at the splice-acceptor site of intron 13: (a1) patient 1 sequence; (a2) reference sequence. (b) Pedigree of family 1; n, normal allele; m, mutated allele. (c) Electropherogram of family 2 with detected heterozygous missense c.998G>A mutation in exon 7 of KCNQ2: (c1) patient 2 sequence; (c2) reference sequence. (d) Pedigree of family 2; n, normal allele; m, mutated allele. (e) Putative canonical splice sites near the c.1526-2A>G mutation (NetGene2 prediction tool). (f) Gene content of the deleted region on 20q13.33 (chr20:63045847-63456499, GRCh38/hg38) in patient 3 based on the Database of Genomic Variants (http://dgv.tcag.ca/dgv/app). The rectangle represents the range of the deletion.
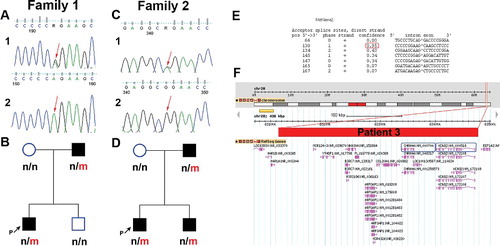
The novel heterozygous splice-site mutation c.1526-2A>G in patient 1, with BFNS, was located at the splice-acceptor site of intron 13. It was inherited from the father, who had had neonatal convulsions in the first days of life that remitted spontaneously. Neither the unaffected family members, nor 100 control individuals carry this mutation ((b)). According to the NetGene2 prediction software, there is 95% confidence for the use of the first alternative AG dinucleotide located three nucleotides downstream in exon 14 ((e)). The consequence might be deletion of one amino acid (c.1526_1528delAAG; p.Glu508del) in the C-terminal domain of the Kv7.2 protein. This domain consists of two coiled-coil motifs known to interact with several modulators of Kv7.2 channel activity, one of which is calmodulin (CaM) Citation[7]. The deletion is located in the second interacting motif (amino acids 501–529) Citation[8]. Another possible consequence is the usage of the splice-acceptor site of some of the adjacent introns, subsequent exon skipping and activation of nonsense-mediated mRNA decay process with resultant non-productive translation. A similar splicing mutation predicted to be pathogenic was found in a neighbouring intron (IVS15-2A>G), in a sporadic case with BFNS Citation[9].
The missense mutation c.998G>A (р.Arg333Gln) in exon 7 was found in patient 2, with primary diagnosis of BFNIS, which later progressed to BECTS. Segregation analysis of family 2 revealed that the missense mutation p.Arg333Gln was also present in the younger brother, who had been diagnosed with BFNS, and the father, who had had neonatal convulsions in his childhood which remitted spontaneously later in life ((d)).
p.Arg333Gln is located in the first CaM-binding motif (amino acids 321–358) of the Kv7.2 C-terminal intracellular domain. KCNQ2 mutants lacking this motif are unable to bind to CaM and do not generate detectable M-currents when co-expressed with KCNQ3 in CHO cells Citation[7,8].
This mutation has been previously reported in a familial case of BFNS Citation[10]. The authors evaluated its effect in the Xenopus oocytes expression system and detected that the rate of deactivation of the channel was much faster for p.Arg333Gln KCNQ2 mutants and the current reduction was relatively lower (<50%) compared to the wild-type channel.
Only three cases of BFNS with point mutations in KCNQ2 who later developed BECTS have been reported so far Citation[3–5]. Two siblings with BFNS progressing to BECTS, having a nonsense mutation (p.Gln323X) located in the same CaM-interacting motif as the mutation in our patient have been described Citation[3]. A point mutation (c.2043delT) in KCNQ2 was reported in an Italian familial case with BFNS, who subsequently developed BECTS during sleep at three years of age Citation[4]. The third case of post-benign neonatal seizures BECTS was reported in a Japanese girl with de novo heterozygous deletion (p.Phe304del) affecting the sixth transmembrane domain of the Kv7.2 protein Citation[5]. The clinical heterogeneity with respect to the seizure onset observed in patient 2 with BFNIS and his relatives with BFNS, as well as in similar BFNS cases reported before, might be due to the influence of additional genetic factors, such as mutation in other epilepsy-associated genes, epigenetic or environmental factors.
In addition, using MLPA analysis, a de novo heterozygous deletion was found in patient 3, covering all 17 exons of KCNQ2. Further aCGH analysis showed that this deletion was 0.41 Mb in size (chr20:63045847–63456499, GRCh38/hg38) and encompassed 11 genes, including two genes associated with epileptic conditions, KCNQ2 and CHRNA4 ((f)).
CHRNA4 encodes alpha-4 neuronal acetylcholine receptor subunit. Missense and insertion mutations in this gene have been associated with autosomal dominant frontal lobe epilepsy (ADNFLE). Deletions restricted only to the KCNQ2 and CHRNA4 genes, similar in size to that in our patient, have been reported before Citation[6,11,12]. The clinical phenotype is a typical BFNS, not presented with the characteristics of ADNFLE. Moreover, α4−/− knockout mice do not exhibit any seizures Citation[13]. Therefore, it is conceivable that this particular phenotype is most likely due to haploinsufficiency of the KCNQ2 gene.
Previously, it has been suggested that deletions encompassing these genes are frequent in developmentally delayed patients with behavioural problems Citation[14]. Nevertheless, deletions of a similar size have been reported in individuals with normal developmental outcome Citation[6,12]. In more severely affected patients having additional neurodevelopmental problems and dysmorphism, there have been described larger deletions ranging from 520 kb to 6.8 Mb in size involving other genes in addition to KCNQ2 and CHRNA4, although the mechanisms leading to atypical clinical features are still unknown Citation[15,16]. Conversely, in patients with smaller size deletions, encompassing KCNQ2 and CHRNA4 genes only, the phenotype is typical BFNS Citation[11,12]. Thus, the size and location of the contiguous gene deletion affecting the whole KCNQ2 gene along with several other genes in the 20q13.33 region in patient 3 is in agreement with the hypothesis that deletions encompassing KCNQ2 and CHRNA4 may not display additional developmental abnormalities Citation[12,16].
Mutations in the KCNQ2 gene are associated with a wide spectrum of electroclinical syndromes with mild to severe outcome. Identification of pathogenic variants in KCNQ2 will clarify and confirm the diagnosis of the affected patient, which will allow more accurate prognosis and subsequent individualized treatment. Furthermore, it will help to estimate the carrier status of relatives at risk, offer prenatal diagnosis and improve the genetic counselling in the affected families.
Conclusions
To our knowledge, this is the first comprehensive study of KCNQ2 in Bulgarian epilepsy patients to reveal three different KCNQ2 mutations associated with BFNS and BFNIS phenotypes. The novel splice-site mutation c.1526-2A>G was found in a patient with typical BFNS phenotype. It enriches the spectrum of KCNQ2 mutations for this particular group of disorders. A contiguous gene deletion affecting the whole KCNQ2 gene along with several other genes in the 20q13.33 region was observed in another patient with typical BFNS. Its size and location suggest that deletions encompassing KCNQ2 and CHRNA4 may not involve additional developmental abnormalities. Finally, a previously reported missense mutation c.998G>A (p.Arg333Gln) associated with BFNS was found in a patient with BFNIS, who subsequently developed BECTS. The observed clinical heterogeneity in this case is a prerequisite for future studies to elucidate the effect of additional genetic factors. The results from the present study will improve the prognosis, treatment and genetic counselling of the affected families.
Acknowledgments
The authors thank all the families participating in the study.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Deprez L, Jansen A, De Jonghe P. Genetics of epilepsy syndromes starting in the first year of life. Neurology. 2009;72(3):273–281.
- Loiseau P, Duché B, Cordova S, et al. Prognosis of benign childhood epilepsy with centrotemporal spikes: a follow-up study of 168 patients. Epilepsia. 1988;29(3):229–235.
- Maihara T, Tsuji M, Higuchi Y, et al. Benign familial neonatal convulsions followed by benign epilepsy with centrotemporal spikes in two siblings. Epilepsia. 1999;40(1):110–113.
- Coppola G, Castaldo P, Miraglia Del Giudice E, et al. A novel KCNQ2 K+channel mutation in benign neonatal convulsions and centrotemporal spikes. Neurology. 2003;61(1):131–134.
- Ishii A, Miyajima T, Kurahashi H, et al. KCNQ2 abnormality in BECTS: benign childhood epilepsy with centrotemporal spikes following benign neonatal seizures resulting from a mutation of KCNQ2. Epilepsy Res. 2012;102(1–2):122–125.
- Zara F, Specchio N, Striano P, et al. Genetic testing in benign familial epilepsies of the first year of life: clinical and diagnostic significance. Epilepsia. 2013;54(3):425–436.
- Wen H, Levitan B. Calmodulin is an auxiliary subunit of KCNQ2/3 potassium channels. J Neurosci. 2002;22(18):7991–8001.
- Yus-Nájera E, Santana-Castro I, Villarroel A. The identification and characterization of a noncontinuous calmodulin-binding site in noninactivating voltage-dependent KCNQ potassium channels. J Biol Chem. 2002;277(32):28545–28553.
- Steinlein O, Conrad C, Weidner B. Benign familial neonatal convulsions: always benign? Epilepsy Res. 2007;73(3):245–249.
- Singh NA, Westenskow P, Charlier C, et al. KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain. 2003;126(12):2726–2737.
- Kurahashi H, Wang J, Ishii A, et al. Deletions involving both KCNQ2 and CHRNA4 present with benign familial neonatal seizures. Neurology. 2009;73(15):1214–1217.
- Okumura A, Ishii A, Shimojima K, et al. Phenotypes of children with 20q13.3 microdeletion affecting KCNQ2 and CHRNA4. Epileptic Disord. 2015;17(2):165–171.
- Marubio LM, del Mar Arroyo-Jimenez M, Cordero-Erausquin M, et al. Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature. 1999;398(6730):805–810.
- Béri-Deixheimer M, Gregoire M, Toutain A, et al. Genotype–phenotype correlations to aid in the prognosis of individuals with uncommon 20q13.33 subtelomere deletions: a collaborative study on behalf of the ‘association des Cytogénéticiens de langue Française’. Eur J Hum Genet. 2007;15(4):446–452.
- Pascual F, Wierenga K, Ng Y. Contiguous deletion of KCNQ2 and CHRNA4 may cause a different disorder from benign familial neonatal seizures. Epilepsy Behav Case Rep. 2013;1:35–38.
- Allen NM, Mannion M, Conroy J, et al. The variable phenotypes of KCNQ-related epilepsy. Epilepsia. 2014;55(9):e99–e105.