
ABSTRACT
The aim of this study was to investigate the bacterial community habituating P18, the biggest crystallizer pond in Pomorie salterns (34% salinity). The obtained results showed that the bacterial community differs from many previous reports of low bacterial diversity in hypersaline environments and demonstrates unusually high diversity of presented taxa, some unusual domination of diverse genera not reported before as dominant and identification of previously unknown 16S rRNA sequences. The retrieved 23 bacterial operational taxonomic units (OTUs) affiliated with 15 bacterial genera from four phyla – Firmicutes, 47.5%; Proteobacteria, 23.1%; Bacteroidetes, 22%; Deinococcus–Thermus, 2.4%; and one-candidate division SR1, 4.8%. Representatives of the phylum Firmicutes predominated in the bacterial community with almost half of the retrieved sequences. Almost all clones branched together with cultured halophiles or uncultured clones retrieved from saline niches. Despite of the high salt concentration, some of the closest phylogenetic neighbours were moderate halophiles. New sequences represented 42.3% of bacterial OTUs. Some of them formed separate branches with similarity less than 85%.
Introduction
Halophilic (‘salt-loving’) microorganisms represent one of the extremophile types living in the most extreme environments on our planet such as salty soils, coastal salterns, salt lakes, underground salt mines and salted foods. Knowledge of the biodiversity in salty environments opens up a possibility to identify novel microorganisms and to understand the functioning of their ecosystems [Citation1]. Additionally, microbial research in saline environments is important for multiple reasons: halophilic microorganisms produce unusual compounds which could be a basis for novel biotechnological applications; terrestrial saline environments may shed light on life on Mars, as this planet is rich in salt deposits and saline environments; primordial life on earth might have started in hypersaline environments [Citation2].
It is well known that molecular techniques have helped to reveal an astonishing level of prokaryotic diversity in environmental samples, while traditional cultivation methods identify only a small fraction of the microorganisms (0.1%–10%) [Citation3]. The use of culture-independent methods for studying prokaryotic diversity based on direct polymerase chain reaction (PCR) amplification of the gene-encoding small-subunit 16S rRNA from environmental DNA samples has led to a significant improvement in the idea of the numbers of existing taxa [Citation4]. The small deal of culturable microorganisms in comparison with the deposited sequences very often hinders setting the firm taxonomic position of the novel sequences [Citation5].
Previous studies on the microbial diversity of hypersaline environments have shown that halophilic members of the domain Bacteria represent a minor fraction in comparison with Archaea representatives [Citation6]. Investigations on the bacterial communities in several saline niches based on 16S rRNA sequences have revealed the presence of common phyla like Cyanobacteria, Proteobacteria, Firmicutes, Actinobacteria, Spirochaetes and Bacteroidetes [Citation7]. Nonetheless, some distinctive microbial communities with different structures have been reported for different geographical location assemblages [Citation8,Citation9].
Until recently, the microbial diversity in halophilic niches in Bulgaria, such as Burgas and Pomorie salterns (PS), and the mine rock saltern in Provadia, was still poorly studied. Recently, we reported on the archaeal diversity in Pomorie salterns [Citation10] and here we describe the results from our investigation on the structure of the other microbial domain of life, Bacteria. Pomorie salterns (42.63N, 27.62E) is located north of the town of Pomorie, at the Bulgarian Black Sea coast. Its area is about 8–8.5 km2, with a length of 5–6 km and width varying from 350 m north to 1.6 km in the middle part and depth not greater than 1.4 m. The temperatures are moderate, with average July temperature of 24 °C and January temperature of 2.7 °C; the average annual rainfall is 598 mm/year.
The aim of this work was phylogenetic characterization of the bacterial community in the biggest crystallizer pond PS 18 (salinity of 340 g/L) from Pomorie saltern using molecular phylogenetic analysis. The diversity in the samples under investigation and that reported for other similar ecosystems was compared.
Materials and methods
Sampling and water analysis
Brine water was collected aseptically in June 2014 from 10 different sites of the crystallizer pond PS18 in order to obtain a representative sample. The homogenized sample was transported in a cooler bag to the lab and was stored at −4 °C prior to initiation of the DNA isolation procedure. The chemical and physical properties of the water sample were analysed by a commercial water chemistry laboratory (DIAL Ltd, Sofia, Bulgaria).
DNA isolation, PCR amplification and construction of 16S rRNA libraries
A sample (3 L) was concentrated by cross-flow filtration through sterile hollow fibre cartridges (1.2-µm pore-size glass fibre prefilter and 0.2-µm membrane filter; Millipore, Bedford, MA, USA). The filter was stored at −20 °C for subsequent DNA extraction. The sample material was suspended in 10 mL of 0.12 mol/L sodium phosphate buffer. Lysis of the cells was achieved after adding sodium dodecyl sulphate (final concentration 2%), NaCl (0.5 mol/L) and polyethylene glycol 6000 (20%). The protocol encompassed three cycles of freezing and thawing, chemical lysis in an extraction buffer (200 mmol/L Tris, pH 8.0), 50 mmol/L ethylenediaminetetra-acetic acid (EDTA), 200 mmol/L NaCl, 2 mmol/L sodium citrate, 10 mmol/L CaCl2–polyadenosine (100 mg/mL)–lysozyme (5 mg/mL), and a proteinase K step. The crude DNA was purified with the AXG-100 Nucleobond cartridges (Machery-Nagel, Düren, Germany) according to the manufacturer's instructions. The eluate was precipitated with 0.7 volumes of isopropanol. The dried pellet was dissolved in Tris-EDTA buffer. The integrity of the DNA was checked by horizontal electrophoresis in 1% agarose (Sigma, Saint Louis, MO, USA) gel in Tris-acetate-EDTA buffer and visualized with ethidium bromide (0.5 mg/L). Samples were extracted in duplicates. All reagents and glassware were autoclaved or filter sterilized before use.
The extracted genomic DNA was used as a target for PCR amplification of 16S rRNA bacterial genes. Community ribosomal DNAs were amplified from 1 to 50 ng of bulk DNA in reaction containing (as final concentrations) 1 × PCR buffer, 2 mmol/L CaCl2, 4 × 200 μmol/L deoxynucleoside triphosphates, 400 nmol/L of each forward and reverse primer, 0.5 U Taq polymerase (GenetBio, Daejeon, Korea). Small-subunit rRNA genes were selectively amplified from the purified community DNA by PCR using two primers universal for the bacterial domain, 8F and 1492R [Citation11], corresponding, respectively, to positions 8–28 and to the complement of the positions 1492–1509 of Escherichia coli 16S rRNA. Reaction mixtures were incubated in BioRad thermal cycler T100 (Foster City, CA, USA) using initial denaturation at 94 °C for 3 min, followed by 30 cycles of 94 °C for 30 s, 55 °C for 30 s and 72 °C for 1 min, and a final extension at 72 °C for 20 min.
PCR products were cloned in E. сoli JM 109 using pJet1.2 cloning kit (Fermentas, Burlington, Ontario, Canada) according to the manufacturer's instructions. Cloned fragments were re-amplified using pJet1.2 forward and reverse primers located in the vector and surrounding the inserted PCR fragment.
Analysis of libraries and clone selection
Screening of the libraries was conducted with two separate restriction fragment length polymorphism (RFLP) analyses. For obtaining the highest resolution of RFLP analysis, four-base restriction enzymes were used. Ten microlitres of the re-amplified PCR products were separately digested with 5 U of each endonuclease, MspI and HaeIII (Fermentas, Burlington, Ontario, Canada) in a final volume of 20 μL for 2 h at 37 °C according to the manufacturer's instructions. The generated fragments were separated in a 2% agarose gel. Restriction fragments shorter than 100 bp were not considered in the analysis. Bands were visualized by staining with ethidium bromide and ultraviolet illumination. The clones with the same restriction patterns were grouped in one operational taxonomic unit (OTU). At least one clone per a restriction pattern was sequenced. The rarefaction curve reflecting the coverage of the diversity was obtained using Analytical Rarefaction version 1.3 (http://www.uga.edu/strata/software/index.html).
16S rRNA gene sequencing and analysis
16S rRNA gene sequences were determined with Applied Biosystems 373A DNA sequencer (Appera Corporation, Foster City, CA, USA) by using the ABI PRISM cycle sequencing kit (Macrogen, Meibergdreef, The Netherlands), where they were re-amplified by using the above primers. 16S rRNA gene sequences were initially compared with reference sequences at National Center for Biotechnology Information (NCBI) database (http://www.ncbi.nlm.nih.gov) using the basic local alignment search tool (BLAST) [Citation12] and Ribosomal Database Project resources [Citation13] to determine their close relatives and approximate phylogenetic affiliations. Phylogenetic analysis was conducted using MEGA version 6.0 [Citation14] and neighbour-joining method [Citation15]. Cloned sequences were checked for possible chimeric structures using the program Chimera Check at the Ribosome Database Project website (http://rdp.cme.msu.edu/html) and the established five chimera sequences were excluded from further analysis. Those of the clones that showed less than 97% similarity to the closest relative after sequencing were referred as new (>3% evolutionary distance of the gene).
The degree of the diversity in the sample was measured with both, cover analysis [Citation16], in which the coverage value was derived from the equation C = 1 − (n/N) × 100, where n is the number of unique clones and N is the total number of the examined clones, and the Shannon index [Citation17]: H = −sumi (pi ln(pi)), where pi is the relative frequency of the ith clone among the examined clones.
Nucleotide sequence accession numbers
The bacterial 16S rRNA gene sequences reported in this study were submitted to the European Molecular Biology Laboratory, GenBank databases under accession numbers LN865054–LN865079.
Results and discussion
Salt composition of water
The analysis of the water from the crystallizer pond P18 showed the following ion composition: 188.38 g/L Cl−, 26.59 g/L SO42−, 101.10 g/L Na+, 18.02 g/L Mg2+, 6.21 g/L K+, 0.32 g/L Ca2+, 0.076 g/L B3+ and 0.022 g/L Sr2+. The total salt concentration was 34%, pH 7.8; the electrical conductivity was 197.6 mS/cm.
16S rDNA library and phylogenetic analysis
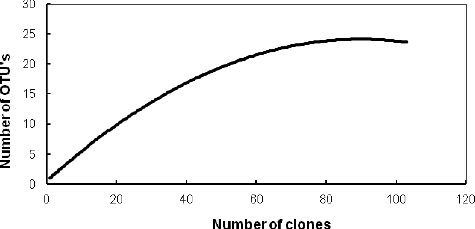
The clone library contained 121 selected clones with inserts of the expected size (approximately 1500 bp). Five of them were not analysed as they showed chimeric structures. The remaining 116 clones were grouped into 23 OTUs after a restriction analysis (). The diversity coverage value C was 95.7%; Shannon's index N was 5.58. Nearly one-fifth of the analysed sequences were singletons, presented only once in the clone library. The abundance of the analysed clones was evaluated by rarefaction analysis (). A decrease in the detection rate of new OTUs was observed with increasing the number of the restricted clones and a clear plateau was reached, suggesting that the main groups presented in the library were detected.
Table 1. Frequencies of OTUs within the bacteria domain derived from the 16S rRNA sequences from а crystallizer pond from Pomorie saltern, Bulgaria*.
Figure 1. Rarefaction curve for sampling of PS 16S rRNA bacterial gene library.
The sequences retrieved from PS were related to 15 bacterial genera referred to four bacterial phyla (): Firmicutes, 47.5%; Proteobacteria, 23.1%; Bacteroidetes, 22%; and Deinococcus–Thermus, 2.4%; and one-candidate phylum, Candidate division SR1, 4.8%. Three of them (Firmicutes, Proteobacteria and Bacteroidetes) are included in the list of six phyla with known halophiles [Citation18]. The unexpected high-number genera differed from the common opinion for low bacterial diversity in most of the hypersaline environments and domination of only one cluster [Citation19,Citation20] and determined this niche as belonging to saltern ecosystems with highest bacterial diversity. It was significantly higher than that observed by other authors in hypersaline salterns with higher than 30% salt content of the sampling site (): three bacterial genera have been established in Maras salterns, Peru [Citation21]; five bacteral genera in a 32% salt pond, Santa Pola salterns, Spain [Citation19]; four bacterial genera in 30% salt ponds, Guerrero Negro saltern, Mexico [Citation22]; and five bacterial genera in the 31% salt pond S5 from a solar saltern in Tunisia [Citation23]. The equal number of genera (15 each) represented in both archaeal [Citation10] and bacterial communities from PS differs from the observations of other authors that members of the domain Archaea are dominant, whereas those of the domain Bacteria are slightly represented in hypersaline environments [Citation4,Citation21–24]. Like our study, Dillon et al. [Citation22] have reported similar archaeal and bacterial 16S rRNA diversity (four genera each) in a hypersaline pond in evaporative Guerrero Negro salterns.
Figure 2. Genus affiliation of 16S rDNAs clones obtained from Pomorie salterns.
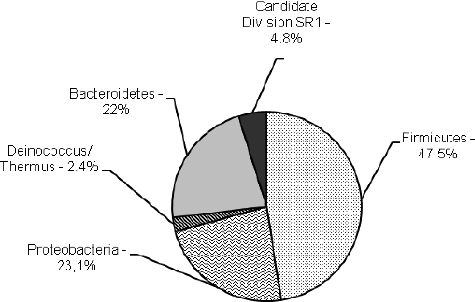
Table 2. Comparison of the microbial diversity in Pomorie saltern compared to other thalassohaline hypersaline ecosystems.
Clones related to Firmicutes represented almost half of all clones (). The domination of Firmicutes in PS is not in agreement with the opinion that Bacteroidetes is the most abundant bacterial phylum in clone libraries from crystallizer ponds [Citation25,Citation26]. According to Antón et al. [Citation8], the presence of different bacterial taxa in them could be due either to their import from previous ponds with lower salinity or to their active growth in the crystallizers. The high number of the clones affiliated with this phylum suggested their active growth in PS. The detected value of 290 mg/L organic carbon in the analysed water could be due to vegetation growing around the salterns, which favours the growth of heterotrophic bacteria from the phyla Firmicutes and Bacteroidetes. Seven of the nine detected Firmicutes OTUs affiliated with representatives of Class Clostridia, with predomination of Halanaerobium clones with 86%–99% similarity to the closest matches. Within the lineages of Firmicutes, we found clones grouped within both the aerobic and anaerobic branches of Class Bacilli: one belonged to the anaerobic genus Anaerobacillus and another one, to the non-spore-forming genus Planomicrobium.
Figure 3. Neighbour-joining phylogenetic tree based on bacterial 16S rDNA sequences found in a crystallizer pond, PS.
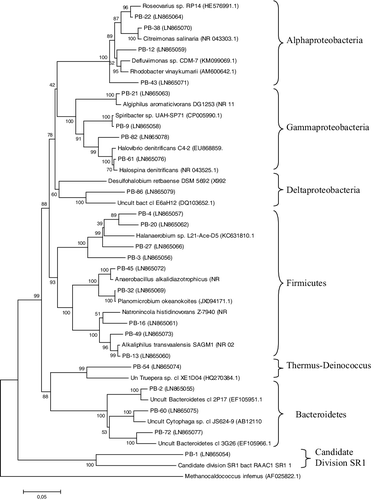
OTUs affiliated with four genera from Class Alphaproteobacteria (Defluviimonas, Roseovarius, Citreimonas and Rhodobacter) and four genera from Gammaproteobacteria (Algiphilus, Halospina, Halovibrio and Spiribacter) were established in the PS library. Some close phylogenetic neighbours for some proteobacterial OTUs were found to be bacterial species isolated from hypersaline environments but with optimal growth at moderate salt concentrations, e.g. Citreimonas salinaria, Roseovarius tolerans, Spiribacter salinus, both Desulfohalobium species [Citation27–31]. An ability of moderate halophilic bacteria to grow in hypersaline habitats has been reported by Oren [Citation18] and probably their classification as moderate halophiles is provisional, as it concerns their ability to grow at such salt concentration in laboratory conditions. Although sequences related to four cultured lineages from Gammaproteobacteria were retrieved in the PS clone library, representatives of the commonly cultivated genera Salicola and Halomonas [Citation32] were not identified. Deltaproteobacterium OTU (PB-86) showed only modest similarity (96%) with an environmental clone found in the Guerrero Negro solar salterns [Citation22] and was not closely related to cultured members of this subphylum (>15% similar to cultured Deltaproteobacterial species). Recovering of sulphate-reducing lineages from Deltaproteobacterium has been already reported for hypersaline environments [Citation22,Citation33].
The third most abundant phylum was Bacteroidetes (22%), whose sequences were spread in three OTUs. The uncultured Bacteroidetes lineage has been reported to comprise 69% of all bacterial 16S rRNA clones from Pond 11 in Guerrero Negro salterns [Citation22] and 50% in the Chula Vista salterns [Citation32]. Although Salinibacter has been found ubiquitously in numerous hypersaline environments [Citation8,Citation34], it was not identified in either PS or in Maras salterns [Citation21]. Abundance of new Bacteroidetes groups, different from Salinibacter, in the salterns has been established by single-cell genome analyses [Citation35]. Phylum Thermus--Deinococcus and Candidate division SR1 were represented by only one OTU each in PS.
Almost all of the bacterial sequences with two exceptions were grouped in the phylogenetic tree with culturable halophiles or unculturable clones from saline niches (mostly hypersaline) worldwide such as coastal salterns and hypersaline lakes. Comparison of the sequences with sequences in databases showed the best matches with unculturable clones corresponding to the hitherto-uncultured members of the domain Bacteria for most of the OTUs (15 of them) (). Only five bacterial OTUs (19%) were affiliated with ≥97% sequence similarity with cultivated species. The closest bacterial matches were recovered from 13 salt lakes or salterns without a clarified similarity to the community composition with clones from a certain niche. Among them, four bacterial OTUs showed the best match with clones from Guerrero Negro solar salterns, Baja CA Sur, Mexico [Citation22]. Pomorie salterns and Guerrero Negro salterns have similar salinity (34%–38%); however, they differ in their very distant geographic locations and in the main cation present in the brine being Na+ for PS and K+ for Guerrero Negro salterns. This result suggests that the type of the ions in salterns has significantly lower influence on the microbial diversity than the degree of salinity.
Degree of novelty of PS bacterial sequences
Almost half of the recovered bacterial sequences (42.3%) were referred as new ones, as they differed with more than 3% from the closest matches. Although the share of new sequences in the bacterial library (42.3%) was less than that in the archaeal one (53.9%) [Citation10], the degree of novelty concerning the higher taxonomic level and sequences with similarity of less than 90% to the closest match and 85% to the culturable neighbour were established. Bacterial diversity on the level of higher taxa could be explained by the reported observation that halophilic bacteria are less mobile or less robust across large distances than halophilic archaea and show a higher tendency towards endemism [Citation36]. Sequences with similarity of less than 90% to the closest match and 85% to the culturable neighbour were established. The similarity with the closest match for five of them was below the empirical limit of 94% sequence identity typically considered to discriminate genera [Citation37]. PB-1 exhibited <85% identity to the known sequences accepted as a boundary for novel bacterial division [Citation38]. The new sequences were distributed in all represented phyla, suggesting that the novelty was not limited to only one phylogenetic branch. Four of the 23 analysed sequences (PB-1, PB-54, PB-72 and PB-86) showed similarity to the closest classifiable relative in databases lower than a certain preset sequence similarity threshold, e.g. 80% [Citation39]. These sequences were referred to four different phyla. The clone PB-1 grouped together with a Candidate division SR1 bacterium RAAC clone isolated from sediment from the Sulphur River in Parkers Cave, Kentucky. The similarity between them was 81%, which was far from the 15% sequence divergence cut-off for a new phylum. The big fraction of novel bacterial and archaeal [Citation10] sequences suggests presence of novel taxa at different levels in the investigated environment. Candidate division SR1, which has been previously included as a part of Candidate division OP11 [Citation40], could be a heterogeneous group and further investigations suppose a division of this phylum.
Conclusions
This study revealed the presence of halophilic bacterial genera that so far had not been considered characteristic for other salterns. The observed impressive bacterial diversity, low similarity with culturable representatives and low match to unculturable clones for many of them is unique for this hypersaline niche and could be explained by the lower degree of knowledge of halophilic bacteria in hypersaline environments. The large fraction of novel bacterial and archaeal sequences suggests that novel taxa at different levels may be present in the investigated environment.
Acknowledgments
The authors thank Dr Ivailo Georgiev (Institute of Microbiology, Bulgarian Academy of Sciences) for his work on the biodiversity indices.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Lizama C, Monteoliva-Sanchez M, Prado B, et al. Taxonomic study of extreme halophilic archaea isolated from the “Salar de Atacama”, Chile. Syst Appl Microbiol. 2001;24:464–474.
- Jiang H, Dong H, Yu B, et al. Microbial response to salinity change in Lake Chaka, a hypersaline lake on Tibetan plateau. Environ Microbiol. 2007;9:2603–2621.
- Hugenholtz P, Goebel BM, Pace NR. Impact of culture independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol. 1998;180:4765–4774.
- Manikandan M, Kannan V, Pašic L. Diversity of microorganisms in solar salterns of Tamil Nadu, India. World J Microbiol Biotechnol. 2009;25:1007–1017.
- Prosser J, Jansson JK, Liu W. Nucleic-acid-based characterization of community structure and function. In: Liu W-T, Jansson JK, editors. Environmental molecular microbiology. Poole: Caister Academic Press; 2010. p. 63–86.
- Ovreas L, Daae FL, Torsvik V, et al. Characterization of microbial diversity in hypersaline environments by melting profiles and reassociation kinetics in combination with terminal restriction fragment length polymorphism (T-RFLP). Microb Ecol. 2003;46:291–301.
- Oren A. Diversity of halophilic microorganisms: environments, phylogeny, physiology, and applications. J Ind Microbiol Biotechnol. 2002;28:56–63.
- Antón J, Rossello MR, Rodríguez-Valera F, et al. Extremely halophilic bacteria in crystallizer ponds from solar salterns. Appl Environ Microbiol. 2000;66:3052–3057.
- Trigui H, Masmoudi S, Brochier-Armanet C, et al. Characterization of heterotrophic prokaryote subgroups in the Sfax coastal solar salterns by combining flow cytometry cell sorting and phylogenetic analysis. Extremophiles. 2011;15:347–358.
- Kambourova M, Tomova I, Boyadzhieva I, et al. Unusually high archaeal diversity in a crystallizer pond, Pomorie salterns, Bulgaria, revealed by phylogenetic analysis. Archaea [Internet]. 2016 [cited 2016 Nov 23]; 2016:7459679. Available from: https://www.hindawi.com/journals/archaea/2016/7459679/
- Weisburg WG, Barns SM, Pelletier DA, et al. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 1991;173:697–703.
- Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol. 1990;215:3389–3402.
- Maidak BL, Larsen N, McCaughey MJ, et al. The ribosomal database project. Nucleic Acids Res. 1994;22:3485–3487.
- Tamura K, Stecher G, Peterson D, et al. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729.
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425.
- Ravenschlag K, Sahm K, Pernthaler J, et al. High bacterial diversity in permanently cold marine sediments. Appl Environ Microbiol. 1999;65:3982–3989.
- Hill TCJ, Walsh KA, Harris JA, et al. Using ecological diversity measures with bacterial communities. FEMS Microbiol Ecol. 2003;43:1–11.
- Oren A. Microbial life at high salt concentrations: phylogenetic and metabolic diversity. Saline Syst [Internet]. 2008 [cited 2016 Oct 18]; 4:2. Available from: https://aquaticbiosystems.biomedcentral.com/articles/10.1186/1746-1448-4-2.
- Benlloch S, López-López A, Casamayor EO, et al. Prokaryotic genetic diversity throughout the salinity gradient of a coastal solar saltern. Environ Microbiol. 2002;4:349–360.
- Burns DG, Camakaris HM, Janssen PH, et al. Combined use of cultivation-dependent and cultivation-independent methods indicates that members of most haloarchaeal groups in an Australian crystallizer pond are cultivable. Appl Environ Microbiol. 2004;70:5258–5265.
- Maturrano L, Santos F, Rosselló-Mora R, et al. Microbial diversity in Maras salterns, a hypersaline environment in the Peruvian Andes. Appl Environ Microbiol. 2006;72:3887–3895.
- Dillon JG, Carlin M, Gutierrez A, et al. Patterns of microbial diversity along a salinity gradient in the Guerrero Negro solar saltern, Baja CA Sur, Mexico. Frontiers Microbiol [Internet]. 2013 [cited 2016 Oct 18];4:399. Available from: http://journal.frontiersin.org/article/10.3389/fmicb.2013.00399/full
- Baati H, Guermazi S, Amdouni R, et al. Prokaryotic diversity of a Tunisian multipond solar saltern. Extremophiles. 2008;12:505–518.
- Boutaiba S, Hacene H, Bidle KA, et al. Microbial diversity of the hypersaline Sidi Ameur and Himalatt salt lakes of the Algerian Sahara. J Arid Environ. 2011;75:909–916.
- Demergasso C, Casamayor EO, Chong G, et al. Distribution of prokaryotic genetic diversity in athalassohaline lakes of the Atacama Desert, Northen Chile. FEMS Microbiol Ecol. 2004;48:57–69.
- Emmerich M, Bhansali A, Lösekann-Behrens T, et al. Abundance, distribution, and activity of Fe (II)-oxidizing and Fe (III)-reducing microorganisms in hypersaline sediments of Lake Kasin, southern Russia. Appl Environ Microbiol. 2012;78:4386–4399.
- Choi DH, Cho BC. Citreimonas salinaria gen. nov., sp. nov., a member of the Roseobacter clade isolated from a solar saltern. Int J Syst Evol Microbiol. 2006;56:2799–2803.
- Labrenz M, Collins MD, Lawson PA, et al. Roseovarius tolerans gen. nov., sp. nov., a budding bacterium with variable bacteriochlorophyll a production from hypersaline Ekho Lake. Int J Syst Bacteriol. 1999;49:137–147.
- León MJ, Fernández AB, Ghai R, et al. From metagenomics to pure culture: isolation and characterization of the moderately halophilic bacterium Spiribacter salinus gen. nov., sp. nov. Appl Environ Microbiol. 2014;80:3850–3857.
- Ollivier B, Hatchikian CE, Prensier G, et al. Desulfohalobium retbaense gen. nov., sp. nov., a halophilic sulfate-reducing bacterium from sediments of a hypersaline lake in Senegal. Int J Syst Bacteriol. 1991;41:74–81.
- Jakobsen TF, Kjeldsen KU, Ingvorsen K. Desulfohalobium utahense sp. nov., a moderately halophilic, sulfate-reducing bacterium isolated from Great Salt Lake. Int J Syst Evol Microbiol. 2006;56:2063–2069.
- Zhaxybayeva O, Stepanauskas R, Mohan NR, et al. Cell sorting analysis of geographically separated hypersaline environments. Extremophiles. 2013;17:265–275.
- Baati H, Guermazi S, Gharsallah N, et al. Novel prokaryotic diversity in sediments of Tunisian multipond solar saltern. Res Microbiol. 2010;161:573–582.
- Makhdoumi-Kakhki A, Amoozegar MA, Kazemi B, et al. Prokaryotic diversity in Aran-Bidgol salt lake, the largest hypersaline playa in Iran. Microbes Environ. 2012;27:87–93.
- Gomariz M, Martínez-García M, Santos F, et al. From community approaches to single-cell genomics: the discovery of ubiquitous hyperhalophilic Bacteroidetes generalists. Int J Syst Evol Microbiol. 2015;9:16–31.
- Pagaling E, Wang H, Venables M, et al. Microbial biogeography of six salt lakes in Inner Mongolia, China, and a salt lake in Argentina. Appl Environ Microbiol. 2009;75:5750–5760.
- Ludwig W, Strunk O, Klugbauer S, et al. Bacterial phylogeny based on comparative sequence analysis. Electrophoresis. 1998;19:554–568.
- Hugenholtz P, Pitulle C, Hershberger KL, et al. Novel division level bacterial diversity in a Yellowstone hot spring. J Bacteriol. 1998;180:366–376.
- Youssef N, Steidley BL, Elshahed MS. Novel high-rank phylogenetic lineages within a sulfur spring (Zodletone spring, Oklahoma, USA) revealed using a combined pyrosequencing/Sanger approach. Appl Environ Microbiol. 2012;78:2677–2688.
- Harris JK, Kelley ST, Pace NR. New perspective on uncultured bacterial phylogenetic division OP11. Appl Environ Microbiol. 2004;70:845–849.