
ABSTRACT
Plasmids are important tools for producing biological reagents and performing molecular biological investigations. They are often constructed by the traditional restriction digestion/ligation cloning method. However, there are some drawbacks of this method due to its nature. It is time-consuming; sometimes there are no suitable restrictive digestion sites and the efficiency of digestion of the polymerase chain reaction (PCR) products is low. To address these problems, we employed an insert cutting-free cloning (ICFC) method in this work. Two kinds of DNA fragments containing the same gene but different terminal sequences were amplified by PCR. Inserts with desired cohesive ends were generated by successively mixing, denaturing and re-annealing these DNA fragments. Finally, they were ligated to the restriction-endonuclease digested vectors. Using this method, we successfully constructed a STAT3 (signal transducer and activator of transcription 3) expression plasmid. The method proved to be simple and efficient. It made the selection of restriction endonucleases and vectors easier, as well as simplified and shortened the cloning process.
Introduction
Plasmid-mediated target gene amplification and over-expression are routine strategies in genetic engineering, biological investigation and industrial production. They have been widely used in the production of many biologically active agents, such as biotechnological enzymes [Citation1–4], medical drugs (such as recombinant interferons, insulins), healthcare products and even foods. What is more, plasmids are one of the simplest but most efficient tools to explore the biological roles of genes/proteins, either by gene overexpression or gene silencing using RNA interference [Citation5–7]. Construction of fusion proteins also allows detecting, localizing and purifying the proteins, as well as investigating the protein–protein interactions more easily [Citation8–10]. Plasmids are also used as carriers of reporter genes to analyze the biological activities of corresponding transcription factors or explore the molecular mechanisms underlying the expression regulation of target genes [Citation11,Citation12]. In addition, site-directed mutagenesis and gene fragmentation are essential in exploring the role of single amino acids or domains in proteins. This also depends on the use of plasmids [Citation13–16].
The traditional restriction enzyme digestion and re-ligation method is a routine way to construct plasmids. However, there are several disadvantages. Firstly, this method relies on restriction enzyme(s)-cutting of the inserts. Therefore, the restriction enzyme cutting sites within the inserts should be avoided, which sometimes leads to failure in selecting suitable restriction enzymes, especially when the aim is to transfer the same inserts into different vectors. Secondly, the low efficiency of digesting the terminal ends of PCR (polymerase chain reaction) products by restriction enzymes often leads to failure of the plasmid construction procedure. The T-A cloning technology can overcome this disadvantage [Citation17]. However, this is a time- and cost-consuming procedure. All these drawbacks result from the requirement of digesting the inserts to generate cohesive ends.
To date, there were several reports of restriction endonuclease digestion-independent cloning methods, including RSFC (restriction site free cloning), SLiCE (seamless ligation cloning extract), SLIC (sequence and ligation-independent cloning), Gibson assembly, CPEC (circular polymerase extension cloning) [Citation18–23]. These methods are effective and reliable. However, they either rely on special enzymes or are too complex.
Here, we introduced a novel method to generate inserts with desired cohesive ends by denaturing and re-annealing two PCR products. Using this insert restriction enzyme cutting-free cloning (ICFC) method, we successfully constructed a pSG5-STAT3 plasmid expressing the signal transducer and activator of transcription 3 (STAT3) gene. This method not only simplified the cloning procedure and saved a lot of time, but also allowed us to select restriction enzymes more easily. It could be widely applied in assembling plasmids.
Materials and methods
Cell lines and reagents
Human leukemia cell line K562 cells (gift from American Type Culture Collection) were cultured in RPMI 1640 (Life Technologies) supplemented with 10% fetal bovine serum (Hyclone, Logan, Utah, USA), 2 mmol/L L-glutamine and antibiotics. African green monkey kidney cell line Cos7 cells were cultured in DMEM (Dulbecco's Modified Eagle's medium; Sigma, St. Louis, MO, USA) supplemented with 10% calf serum (Hyclone), 2 mmol/L-glutamine and antibiotics. Human IFN-α was purchased from Schering-Plough Co. and stored at 4 °C. It was used at a final concentration of 1000 U/mL.
Reverse transcription (RT)-PCR
K562 cells (5 × 106) were collected and total RNA was extracted with Trizol reagent (Invitrogen, Carlsbad, CA, USA). After reverse transcription with 2 μg RNA, Random primer 9 (TaKaRa, Dalian, China), M-MLV reverse transcriptase (Promega, Madison, WI, USA) and cDNA were amplified respectively with PrimeSTAR DNA polymerase (TaRaKa) under the following conditions (Eppendorf 5331 Gradient MasterCycler, Hamburg, Germany): 30 cycles of 98 °C for 10 s, 68 °C for 150 s and 68 °C for 10 min. The primers were STAT3-F1/STAT3-R1 or STAT3-F2/STAT3-R2 ().
Table 1. Primer sequences used in the study.
Generation of plasmid
To construct the plasmid, 500 ng of pSG5 vector (Stratagene, La Jolla, CA, USA) was digested with EcoRI and BglII restriction endonucleases (TaKaRa). The cleaved vector and the two RT-PCR products were separated by 1% agarose gel electrophoresis and purified by Gel Extraction Kit (TaKaRa). The two extracted PCR products were then combined at a mole ratio of 1:1, denatured (94 °C for 5 min) and then re-annealed (50 °C for 2 min). Then the mixture was ligated to the pSG5 vector extracted from the gel using T4 ligase (TaKaRa) overnight at 4 °C. The pSG5 vector/STAT3 gene mixture was transformed into Escherichia coli DH5α competent cells (TaKaRa). The clones were picked and incubated in liquid LB (Luria–Bertani) medium containing 100 μg/mL ampicillin at 37 °C with shaking. Then the plasmids were extracted and verified by both restriction endonucleases cleavage and DNA sequencing (Sanger sequencing method; Shanghai Sangon Biological Engineering, Shanghai, China).
Transfection
The Cos7 cells were cultured in 24-well plates to 30%–40% confluence before transfection. Then they were transfected with the indicated plasmids or empty vectors using Lipofect Transfection reagent (Tiangen Biotech, Beijing, China), according to the manufacturer's instructions. Twenty-four hours after the transfection, the cells were treated with or without IFN-α for 18 hours followed by transcriptional activity assay or protein extraction.
Western blot
Cos7 cells were harvested using a scraper 48 hours after transfection and protein was extracted using Tris-HCl lysis buffer (50 mmol/L Tris-HCl pH8.0, 150 mmol/L NaCl, 0.5% NP-40, 1 × protease inhibitor cocktail (Roche, Switzerland), 100 μg/mL PMSF (phenylmethanesulphonyl fluoride)). The protein extract was quantitated using a BCA (bicinchoninic acid assay) kit (Sangon, Shanghai, China); 5 mg protein extract was separated by 8% sodium dodecyl sulfate–polyacrylamide electrophoresis (SDS–PAGE) and then blotted onto Hybond-C Extra membranes (Amersham Pharmacia Biotech, Uppsala, Sweden). The membrane was blocked with 5% nonfat milk and successively incubated with indicated primary antibodies and horseradish peroxidase (HRP)-conjugated rabbit anti-mouse monoclonal secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The immunocomplex was visualized by an ECL kit (Cell Signaling Technology, Danvers, MA, USA). Primary antibodies were purchased from Santa Cruz Biotechnology (anti-STAT3) and Cell Signaling Technology (anti-β-actin), respectively.
Luciferase reporter assay
The Cos7 cells were cotransfected with the 4 × APREs (acute phase response elements)-luciferase reporter gene construct (gift from Dr. Yuzuru Kanakura, Department of Hematology and Oncology, Osaka University Graduate School of Medicine, Japan) [Citation24] and empty vector/expression plasmid as indicated. Twenty nanograms of pRL-TK vector (Promega) were also cotransfected into the cells to normalize the transfection efficiency. Twenty-four hours after the transfection, the cells were treated with or without IFN-α as indicated for another 18 hours. The transcriptional activity was analyzed using Dual-Luciferase Assay System (Promega), according to the manufacturer's instruction. Data are shown as means with standard deviation (±SD) of at least three independent experiments.
Results and discussion
Preparation of STAT3 gene with desired sticky ends
According to the pSG5 vector map ((A)), there are only three restriction sites in the multiple cloning sites (MCS), EcoRI, BamHI and BglII. By analyzing the sequence of the STAT3 gene coding region, we found all these enzyme cutting sites ((B)), which limited the use of the traditional restrictive enzyme cutting/re-ligation cloning method to construct the plasmid pSG5-STAT3. Here, we employed the ICFC method. We denatured and re-annealed two PCR products to generate the desired sticky ends.
Figure 1. Schematic diagram of pSG5 vector and generation of STAT3 genes with desired cohesive ends. (A) Schematic representation of the pSG5 vector. There are three restrictive cleavage sites, EcoRI, BamHI and BglII, in multiple cloning sites. The recognition sequences and cutting model of the three enzymes are shown (lower panel). (B) Partial coding region sequences of the STAT3 gene. The initiation and termination codons are underlined. Digestion sites of the three restriction enzymes in the STAT3 gene are boxed. (C) Schematic illustration of generating STAT3 genes with desired cohesive ends independent of restriction endonuclease digestion. STAT3 coding-region sequences are shown as grey lower case letters, and cleaving-related sequences of restriction endonucleases, as black uppercase letters. The STAT3 genes with desired cohesive ends are boxed. (D) Agarose gel electrophoresis of the STAT3 gene amplified from K562-cell total RNA by RT-PCR with two pairs of primers. DNA ladder (λ/Hind III DNA Marker, TaKaRa, Dalian, China).
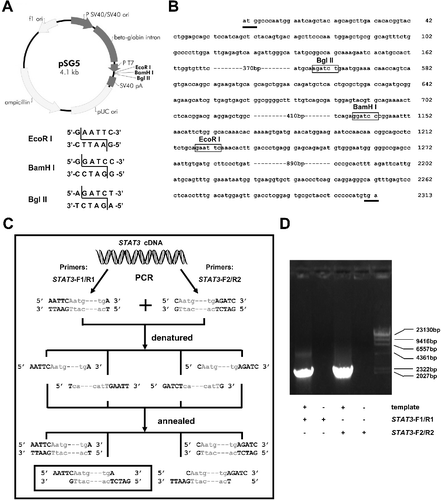
Firstly, EcoRI and BglII were selected as the cloning restriction enzymes. According to the sequences of the sticky ends of the EcoRI and BglII cutting sites ((A)), two pairs of primers were designed and synthesized (STAT3-F1/R1 and STAT3-F2/R2) () and used respectively to amplify the STAT3 gene by RT-PCR ((C) and 1(D)). Then, the two PCR products were successively mixed, denatured and annealed. Hence, four STAT3 amplicons with different terminal ends were assembled (two with blunt ends and two with cohesive ends) ((C)). One of them (sequences with a frame, (C)) was the desired one and also the only one that could be ligated to the EcoRI and BglII-digested pSG5 vector.
Using this method, we did not need to digest the insert to generate the desired sticky ends, so restriction endonucleases could be easily selected according to the restriction sites in the MCS of the target vector. Thus, the proposed method could be widely used. The procedure is also simple. Although one more PCR should be carried out, it did not take a lot of time and efforts. Because the similarity of the two pairs of primers was high, the two PCRs could be carried out simultaneously under the same PCR conditions.
Construction of the pSG5-STAT3 expression plasmid
To construct the expression plasmid pSG5-STAT3, the vector pSG5 was cut by both EcoRI and BglII restriction endonucleases ((A)), and was separated by agarose gel electrophoresis. The gel extraction product of the pSG5 vector was then ligated with the re-annealed STAT3 gene mixture, followed by being transformed into DH5α competent cells. After 18 hours’ culture, 15 clones grew on LB solid medium. Four of them were picked and cultured in LB lipid medium. The plasmids were purified and preliminarily verified by EcoRI restriction endonuclease digestion. As shown in (B), cleavage of the pSG5-STAT3 plasmids with EcoRI would generate two DNA fragments, one 1.2 kb and the other 5.2 kb. The results from the electrophoretic analysis were in accordance with our expected results ((C)). The sequence of the insert of pSG5-STAT3 was further verified by DNA sequencing. Part of the results are shown in (D).
Figure 2. Construction and verification of pSG5-STAT3 plasmid. (A) Digestion of the pSG5 vector by EcoRI and BglII restriction enzymes and separation by agarose gel electrophoresis. (B) Schematic illustration of part of the restriction enzyme cutting sites of the pSG5-STAT3 plasmid. (C) Verification of the pSG5-STAT3 plasmid by restriction endonuclease digestion. Four clones of pSG5-STAT3 plasmids were digested with EcoRI and analyzed by agarose gel electrophoresis. DNA ladders (DL2000 DNA marker (left) and λ/Hind III DNA marker (right), TaKaRa). (D) DNA sequencing results of the insert–vector joints part of the pSG5-STAT3 plasmid (clone 1). The restriction enzyme cutting sites are boxed. The initiation and termination codons are underlined.
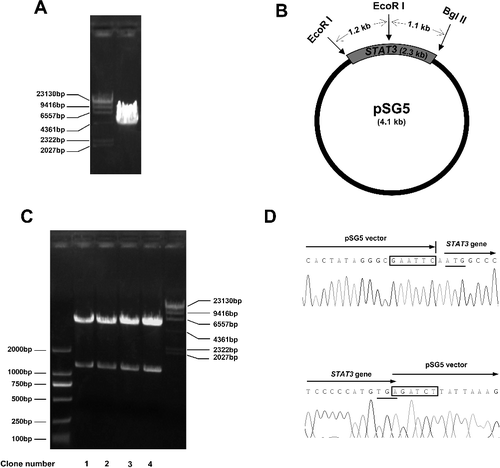
Here, the question was whether there were plasmids with inverted inserts. According to the principle of complementary base pairing, only one of the four potential re-annealed STAT3 inserts could be ligated to the digested pSG5 vector, which is also what we aimed at. In accordance with this, all the four clones in this work turned out to be correct, indicating that this method was reliable and efficient.
Analysis of the expression of pSG5-STAT3
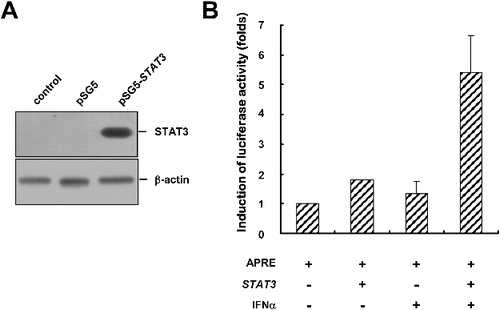
To examine the expression of pSG5-STAT3, Cos7 cells were transfected with the pSG5 empty vector or the pSG5-STAT3 plasmid. The protein level of STAT3 was analyzed by western blot. As shown in (A), no STAT3 protein was detected in the control cells or the cells transfected with an empty pSG5 vector. However, the cells transfected with pSG5-STAT3 showed an obvious increase in STAT3 protein level, indicating pSG5-STAT3 could express the STAT3 protein. Then, an APRE (acute phase response element)-luciferase construct was used to verify the biological function of this exogenous STAT3 protein [Citation24]. The APRE-luciferase reporter gene construct contains four APREs and can respond to IFN-α-induced tyrosine-phosphorylated STAT3 [Citation24–28]. As shown in (B), transfection of Cos7 cells with pSG5-STAT3 did not enhance the luciferase activity, which resulted from the inactivated state of STAT3 (tyrosine 705 was unphosphorylated). However, treatment of STAT3-transfected cells with IFN-α for 18 hours led to an increase in luciferase activity by up to fivefold, suggesting that the exogenous STAT3 protein had biological activity. This indicated the success of using ICFC method to construct plasmids.
Figure 3. Verification of the expression of pSG5-STAT3. (A) Western blot analysis of the expression of the pSG5-STAT3 plasmid in Cos7 control cells (without transfection) or cells transfected as indicated. The expression level of β-actin was used as a loading control. (B) Luciferase reporter assay verification of the biological activity of STAT3, 18 hours after IFN-α treatment.
The ICFC strategy was shown to be an efficient and simple method. It not only made the selection of restriction endonucleases and vectors much easier, but also greatly simplified and shortened the cloning process. It could be applied in almost all cases. This would also be useful for constructing plasmids with large inserts. In this case, gene segments with sticky ends are synthesized respectively and then the large inserts are assembled by ligating these segments.
Conclusions
We designed an insert restriction enzyme cutting-free cloning (ICFC) strategy and successfully constructed a STAT3 expression plasmid pSG5-STAT3. In this way, it was not needed to digest the inserts to generate the desired sticky ends. Thus, it was easy to select restriction endonucleases, regardless of whether their cutting sites are present within the coding sequence of interest. This method could be widely used. It is also simple, time-saving and reliable.
Acknowledgments
We thank all members of the Institute of Hematology of Zhejiang University for their support and help. We also thank Jun Chen (Department of General Surgery, the First Affiliated Hospital of Zhejiang University) for his help.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Mead DA, McClary JA, Luckey JA, et al. Bst DNA polymerase permits rapid sequence analysis from nanogram amounts of template. Biotechniques. 1991;11:76–78.
- McMullan G, Christie JM, Rahman TJ, et al. Habitat, applications and genomics of the aerobic, thermophilic genus Geobacillus. Biochem Soc Trans. 2004;32:214–217.
- Vellore J, Moretz SE, Lampson BC. A group II intron-type open reading frame from the thermophile Bacillus (Geobacillus) stearothermophilus encodes a heat-stable reverse transcriptase. Appl Environ Microbiol. 2004;70:7140–7147.
- Garg N, Tang W, Goto Y, et al. Lantibiotics from Geobacillus thermodenitrificans. Proc Natl Acad Sci USA. 2012;109:5241–5246.
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553.
- Lou YJ, Pan XR, Jia PM, et al. RIG-G inhibits the proliferation of NB4 cells and propels ATRA-induced differentiation of APL cells. Leuk Res. 2016;40:83–89.
- Zhuang LK, Xu GP, Pan XR, et al. MicroRNA-181a-mediated downregulation of AC9 protein decreases intracellular cAMP level and inhibits ATRA-induced APL cell differentiation. Cell Death Dis. 2014 [cited 2017 Jul 04];5:e1161. DOI:10.1038/cddis.2014.130
- Chien CT, Bartel PL, Sternglanz R, et al. The two-hybrid system: a method to identify and clone genes for proteins that interact with a protein of interest. Proc Natl Acad Sci USA. 1991;88:9578–9582.
- Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246.
- Chalfie M, Tu Y, Euskirchen G, et al. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805.
- Lou YJ, Zhang ZL, Pan XR, et al. Intact JAK-STAT signaling pathway is a prerequisite for STAT1 to reinforce the expression of RIG-G gene. Exp Cell Res. 2011;317:513–520.
- Lou YJ, Pan XR, Jia PM, et al. IRF-9/STAT2 [corrected] functional interaction drives retinoic acid-induced gene G expression independently of STAT1. Cancer Res. 2009;69:3673–3680.
- Sugimoto M, Esaki N, Tanaka H, et al. A simple and efficient method for the oligonucleotide-directed mutagenesis using plasmid DNA template and phosphorothioate-modified nucleotide. Anal Biochem. 1989;179:309–311.
- Vandeyar MA, Weiner MP, Hutton CJ, et al. A simple and rapid method for the selection of oligodeoxynucleotide-directed mutants. Gene. 1988;65:129–133.
- Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci USA. 1985;82:488–492.
- Taylor JW, Ott J, Eckstein F. The rapid generation of oligonucleotide-directed mutations at high frequency using phosphorothioate-modified DNA. Nucleic Acids Res. 1985;13:8765–8785.
- Zhou MY, Clark SE, Gomez-Sanchez CE. Universal cloning method by TA strategy. Biotechniques. 1995;19:34–35.
- Vogl T, Ahmad M, Krainer FW, et al. Restriction site free cloning (RSFC) plasmid family for seamless, sequence independent cloning in Pichia pastoris. Microb Cell Fact. 2015 [cited 2017 Jul 04];14:103. DOI:10.1186/s12934-015-0293-6
- Li MZ, Elledge SJ. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat Methods. 2007;4:251–256.
- Zhang Y, Werling U, Edelmann W. SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res. 2012 [cited 2017 Jul 04];40:e55. DOI:10.1093/nar/gkr1288
- Gibson DG, Young L, Chuang RY, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345.
- Quan J, Tian J. Circular polymerase extension cloning of complex gene libraries and pathways. PLoS One. 2009 [cited 2017 Jul 04];4:e6441. DOI:10.1371/journal.pone.0006441
- Dortay H, Akula UM, Westphal C, et al. High-throughput protein expression using a combination of ligation-independent cloning (LIC) and infrared fluorescent protein (IFP) detection. PLoS One. 2011 [cited 2017 Jul 04];6:e18900. DOI:10.1371/journal.pone.0018900
- Kawasaki A, Matsumura I, Kataoka Y, et al. Opposing effects of PML and PML/RAR alpha on STAT3 activity. Blood. 2003;101:3668–3673.
- Mahony R, Gargan S, Roberts KL, et al. A novel anti-viral role for STAT3 in IFN-alpha signalling responses. Cell Mol Life Sci. 2017;74:1755–1764.
- Dixon BJ, Chen D, Zhang Y, et al. Intranasal administration of interferon beta attenuates neuronal apoptosis via the JAK1/STAT3/BCL-2 pathway in a rat model of neonatal hypoxic-ischemic encephalopathy. ASN Neuro. 2016 [cited 2017 Jul 04];8:1759091416670492. DOI:10.1177/1759091416670492
- Ren M, Wang Y, Wu X, et al. Curcumin synergistically increases effects of beta-interferon and retinoic acid on breast cancer cells in vitro and in vivo by up-regulation of GRIM-19 through STAT3-dependent and STAT3-independent pathways. J Drug Target. 2017;25:247–254.
- Xu F, Song H, Li N, et al. HBsAg blocks TYPE I IFN induced up-regulation of A3G through inhibition of STAT3. Biochem Biophys Res Commun. 2016;473:219–223.