
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Lipase is one of the most important industrial enzymes, widely used in the preparation of food additives, cosmetics and pharmaceuticals. In order to obtain a large amount of lipase, in the present study, a gene encoding intracellular lipase was cloned from Acinetobacter haemolyticus. The recombinant lipase KV1 containing a His-tag was expressed in Esherichia coli BL21 (DE3) cells, using pET-30a as the expression vector. Using the central composite design, screening and optimization of induction conditions (cell density before induction, IPTG (isopropyl β-D-1-thiogalactopyranoside) concentration, post-induction temperature and post-induction time) were made. All parameters significantly (P < 0.05) influenced the expression of lipase KV1, rendering a 70% increase in enzyme production at optimum induction conditions (OD600 before induction: 0.6, IPTG concentration: 0.5 mmol/L, post-induction temperature: 40 °C, post-induction time: 16 h). The expressed recombinant lipase KV1 was purified using Ni-affinity chromatography, affording ∼3.1-fold of the enzyme with an estimated relative molecular mass of 39 kDa. The recombinant lipase KV1 exhibited its maximum activity at 40 °C and pH 8.0. Beneficially, the recombinant lipase KV1 retained its relative activities (>80%) even up to 24 h between pH 7−12; suggesting that the recombinant lipase KV1 may be suitable for a wide range of industrial applications.
Introduction
There is a growing demand for industrial enzymes, especially of microbial origin, owing to their wide versatility. Enzyme mediated reactions are considered as less labour-intensive and more cost-effective alternatives to the chemical methods [Citation1]. More industrial players are switching over to the use of enzymes for an array of applications on an industrial scale [Citation2]. Among such enzymes, lipases are preferred in organic chemistry, the pharmaceutical industry, biophysics, biochemical and process engineering, biotechnology, microbiology, biochemistry, etc. The use of lipases (triacylglycerol lipases E.C. 3.1.1.3) as biocatalysts is particularly useful due to their substrate specificity, ability to function in extreme and mesophilic environments, as well as easier bulk production [Citation3].
The interest in Acinetobacter lipase is gradually increasing concomitant with the growth of the enzyme industry, as well as the widening search for novel enzymes and applications [Citation3]. In the present study, the purified wild-type lipase KV1 (accession number: KX459517) that we recently isolated from A. haemolyticus showed maximum relative activity at 40 °C and pH 8.0. Remarkably, the wild lipase KV1 showed extended stability up to 25 h when incubated over a wide range of temperatures and pH, retaining relative activities >80%. Due to high demand for functional bacterial lipases showing neutral or alkaline pH optimum [Citation3,Citation4], overexpressing the wild-type lipase KV1 protein in a suitable bacterial system would increase its quantity for characterization and development for prospective manufacturing uses, as well as biodegradation and bioremediation purposes.
Technologies have been developed over the last few years to increase the production of recombinant proteins to cater various applications, e.g. bioinsecticides, diagnostic and bioremediation processes, especially for the production of detergent and food processing [Citation5]. For initial screening of recombinant protein expression, Escherichia coli is largely the host of choice, since the cells are prone to manipulation and are inexpensively and rapidly grown in culture. However, several important challenges concerning protein solubility and stability prevail, often resulting in the accumulation of the overexpressed proteins as inclusion bodies [Citation6]. Therefore, in the present study, we chose E. coli strain BL21 (DE3) to avoid the above-mentioned problems. The chosen E. coli strain BL21 (DE3) is very advantageous because it has deficiency in proteases Lon and OmpT and can increase the stability, solubility as well as the expression of the protein. Another merit of the strain of interest is the possession of a chromosomal copy of the T7 RNA polymerase gene which supports simple and efficient expression of genes under control of the T7 promoter [Citation6]. The presence of Tags can also improve the affinity of purification, providing easy isolation of the highly pure E. coli proteins, and overexpression of the proteins can be optimized by varying the expression conditions viz. temperature, type of cultivation media and the type of E. coli strain [Citation7]. However, not all proteins can be produced in E. coli due to the heterogeneity of proteins. Heterogeneous protein expression in E. coli can be quite problematic due to failure of the overexpressed recombinant proteins to reach their correct conformation [Citation8]. This problem has been attributed to the lack of necessary post-translational modifications, folding modulators or poor control of the folding rates of large proteins in the tightly packed E. coli cytoplasm [Citation8]. Yet, no method has been universally successful to overcome this problem.
The current conventional one factor-at-a-time approach for optimizing overexpressing recombinant proteins is laborious typically due to the large number of factors and their interactions that can affect expression [Citation9,Citation10]. Such challenges in optimizing protein overexpression may be overcome using statistical approaches like response surface methodology (RSM). Therefore, RSM was used in this study to optimize the cloning and overexpression of the wild-type lipase KV1 protein in E. coli to produce high yields of functional recombinant lipase KV1 protein. Central composite design (CCD) was selected, as this design has been proven successful by several studies for optimizing the overexpression of other lipases [Citation11,Citation12]. The overexpression of recombinant protein can be influenced by multiple parameters, viz. expression in host strain as well as expression conditions such as temperature, inducer concentration, pH, induction time and the composition of the culture medium [Citation8]. Therefore, optimizing the condition parameters using CCD is may prove highly beneficial due to the limited number of experiments required to optimize the overexpression of the recombinant lipase KV1 for high enzyme activity [Citation11].
The aim of this study was to use RSM to optimize the overexpression of A. haemolyticus lipase KV1 in E. coli. The overexpressed recombinant lipase KV1 protein was then purified for subsequent biochemical characterizations. The recombinant lipase KV1 from A. haemolyticus showed various beneficial biochemical properties such as broad substrate specificity, stereo-selectivity, remarkable stability over a broad range of pH values (pH 7−11), as well as an optimum activity at 40 °C. Such characteristics indicated that it was an excellent enzyme for producing a wide range of industrial detergents, cleaning up enviro-agro-industrial wastes, as well as catalysis in synthetic manufacturing processes. Hence, the overexpression of this exceptional lipase protein could be of great importance for future experimental and industrial applications.
Materials and methods
Bacterial strain, plasmid and chemicals
E. coli strains JM109 (Promega, USA) and BL21 (DE3) (Promega, USA) were maintained at 37 °C in Luria–Bertani (LB) broth or on agar plates for recombinant plasmid amplification and protein expression, respectively. The vectors pGEM-T Easy (Promega, Madison, WI, USA) and pET-30a (+) (Promega, USA) were used for gene cloning and expression, respectively. DNA purification kits, restriction endonucleases, T4 DNA ligase and Taq DNA polymerase were all purchased from Promega (USA). Acetic acid, copper (II) acetate-1-hydrate, isooctane, ammonium sulphate, boron, copper, manganese, molydenum, zinc, sodium chloride and methanol were purchased from Merck (Germany). Bovine serum albumin (BSA), Bradford reagent, agarose, triolein, tributyrin, Victoria blue, rhodamine B, nutrient broth and nutrient agar were obtained from Sigma–Aldrich (USA). Arabic gums, magnesium sulphate heptahydrate, magnesium chloride hexahydrate, calcium chloride dihydrate, potassium phosphate monobasic, isopropyl β-D-1-thiogalactopyranoside (IPTG) were purchased from Fisher Scientific (UK). The chemicals used were of analytical grade and are commercially available.
Cloning of the lipase KV1 gene
The purified DNA encoding lipase KV1 was cloned with the pGEM-T-easy vector, according to the manufacturer's instructions, transformed into E. coli JM109 competent cells and subsequently plated on an LB agar plate (100 μg/mL ampicillin). Positive white clones were first screened for lipase activity using a tributyrin-LB agar plate (100 μg/mL ampicillin) and further streaked onto triolein and rhodamine-LB agar (100 µg/mL ampicillin). The plates were incubated at 37 °C for 18 h for the growth of positive transformants [Citation13].
Construction of expression vector of lipase gene
Lipase gene with Nco1 and Xho1 sites was obtained by polymerase chain reaction (PCR) using the forward primer: 5′-TAAGCACCATGGATGACACAACAATCGAGCATGCAC-3′ and the reverse primer: 5′-TGCTTACTCGAGGAACATTGGCTTTAACGAACTCAGC-3′. The reactions were conducted on a Corbett Research FTS-960 thermal cycler. Cycling conditions for the 30 cycles were: 94 °C for 30 s, 55 °C for 45 s, 72 °C for 45 s and 72 °C for 10 min. The PCR products and the vector pET-30a were digested with Nco1 and Xho1, recovered through agarose gel electrophoresis, and then ligated by T4 DNA ligase. The ligated plasmid pet30a-lipKV1 was transformed into E. coli BL21 (DE3). A single colony of the transformants was selected and transferred into a 5 mL LB broth with 100 μg/mL kanamycin, and incubated overnight with vigorous shaking (200 rpm) at 37 °C. The recombinant plasmid was extracted using QIAprep® spin miniprep kit (Qiagen, Germany) according to the manufacturer's instructions [Citation14]. The plasmid was digested with Nco1 and Xho1 in order to identify if the recombined plasmid had been constructed.
Experimental design and optimization for lipase KV1 expression in E. coli
RSM was used to optimize the culture conditions for lipase KV1 expression. A four-factor-five-level CCD that required 30 experiments was used to evaluate the overexpression lipase KV1 [Citation15]. The CCD consisted of 16 factorial points, 8 axial points and 6 centre points. The software package Design Expert version 7.1.2® (Stat-Ease Inc., Minneapolis, USA) was used to investigate the interactive effect of the four relevant variables viz. intensity of the optical density at 600 nm (OD600) before induction (0.4, 0.5, 0.6, 0.7 and 0.8); IPTG concentration (0.1, 0.3, 0.5, 0.7 and 0.9 mmol/L); post-induction temperature (24, 32, 40, 48 and 56 °C) and post-induction time (4, 10, 16, 22 and 28 h). illustrates the minimum and maximum levels of variables chosen for trials in the CCD, while the CCD at the given range of the variables assessed in terms of codes and actual terms is presented in . To optimize the production of lipase KV1 in E. coli, the expression host BL21 (DE3) was used. Cells transformed with pet30a/lipase KV1 were grown at mid-log phase (OD600 of 0.4–0.8 nm) and the expression of the protein was induced with 0.1–0.9 mmol/L IPTG at 24−56 °C for 4–28 h. The intracellular fractions were obtained by centrifuging the culture (10 mL) at 10,000 × g for 10 min at 4 °C. The pellet was resuspended with an equal volume of 50 mmol/L of potassium phosphate buffer (pH 7.0) prior to sonication (Branson 250 sonifier: output 2, duty cycle 30 and min 2) and cleared by centrifugation (12,000 × g, 20 min). The clear crude lysate was used to measure the intracellular lipase activity. All fractions were analysed in a 12% (w/v) sodium dodecyl sulphate (SDS) polyacrylamide gel electrophoresis (SDS-PAGE) to estimate the expression level of the recombinant lipase KV1 [Citation15,Citation16].
Table 1. Coded values of variables used in central composite rotary design.
Table 2. Compositions of the various runs of the CCD in coded and actual terms for the obtained actual and predicted responses for optimizing lipase KV1 activity.
Statistical analysis for RSM
To evaluate the effect of factors on the response surface in the region of investigation, a four-factor-five-level CCD was performed. The ranges and levels of the four variables (A: OD600nm; B: IPTG concentration, C: post-induction temperature and D: post-induction time) are provided in . The experimental data from the CCD ( and ) were analysed by the response surface regression process using the following second-order polynomial Equationequation(1)
(1) :
(1)
(1) where Y is the measured response variable, bo, bi, bi, and bii are constant and regression coefficients of the model with xi and xj represent the independent variables in coded values. The generated second-order polynomial coefficients were used to estimate the response of the dependent variable. Analysis of variance (ANOVA) was carried out to fit the second-order polynomial equations for all response variables. While F-test was used to evaluate the significance of the model equation and model terms, the lack of fit, coefficient of determination (R2), the predicted R2, adjusted R2 and adequate precision were used to express the quality of fit of the polynomial model. The fitted polynomial equations were expressed as three-dimension surface plots to demonstrate the relation between the response experimental levels of each variable used in the CCD. Subsequently, a point optimization process was used to optimize the level of each variable to achieve the maximum response of lipase activity (U/mL). The combination of different optimized variables which contributed to a maximum response [Citation17] was tested experimentally to investigate the viability of the model.
Lipase activity assay and determination of protein content
The activity assay was carried out at 40 °C for 30 min at 200 rpm using olive oil as the substrate, and lipase activity was estimated by the amount of liberated free fatty acid. The released free fatty acid was extracted with isooctane, stained with copper reagent and read at 715 nm using a spectrophotometer (HITACHI U-3210) with isooctane as blank. One unit (U) of lipase activity is defined as the amount of enzyme releasing 1 μmol of fatty acid per minute. In this study, the lipase activity was represented as percentage relative activity. The protein content was estimated from the standard curve prepared using BSA standard solutions and Bradford reagent, monitored at 595 nm in a spectrophotometer (HITACHI U-3210) with solution without BSA as the blank [Citation18]. All determinations were carried out in triplicates.
Purification of the recombinant lipase KV1
The 6xHis-tagged recombinant lipase KV1 was purified using nickel nitrilotriacetic acid (Ni-NTA) sepharose affinity chromatography. The concentrated recombinant lipase enzyme was subjected to Ni-NTA affinity chromatography column (GE Healthcare Life Sciences, USA) pre-equilibrated with washing buffer (50 mmol/L Tris–Cl pH 8.0, 500 mmol/L NaCl and 10% glycerol). The protein was eluted using an imidazole step gradient (0, 20, 40, 80, 100 and 200 mmol/L) in washing buffer. Finally, the imidazole in the purified protein was removed by dialysis in 50 mmol/L Tris–Cl at pH 8.0. The purified recombinant lipase KV1 was evaluated by 12% SDS-PAGE and Western blot analysis [Citation19].
SDS-PAGE and Western blot analysis
The molecular weight of the purified enzymes was determined by 12% SDS–PAGE [Citation20] and the proteins were subsequently transferred onto nitrocellulose membranes (Amersham Pharmacia Biotech,UK)). The membranes were blocked with 2% non-fat milk in phosphate buffered saline, incubated with a His-Tag antibody (QIAGEN) overnight at 4 °C, washed and further incubated with the appropriate secondary antibody coupled to horseradish peroxidase (Amersham Pharmacia Biotech) for 1 h. Immunoblots were developed using the 1-Step Ultra TMB-Blotting Solution (ThermoFisher Scientific).
Characterization of the purified recombinant lipase KV1
Using a standardized protein concentration (1.98 mg/mL), characterization of the relative activity of the purified recombinant KV1 lipase was performed.
Effect of temperature on lipase activity and stability
The effect of temperature on the relative activity of recombinant KV1 lipase was determined at temperatures ranging from 5 to 80 °C for 30 min, prior to the activity assay. Using the same temperature range (5−80 °C), the enzyme stability test was conducted by pre-incubating the lipase for 30 min and the stability was monitored for 24 h [Citation21].
Effect of pH on lipase activity and stability
The effect of pH was evaluated using various buffer systems at 200 rpm for 30 min, viz. 50 mmol/L acetate buffer (pH 4−6), potassium phosphate buffer (pH 6−8), Tris–Cl buffer (pH 8−9), glycine–NaOH (pH 9−11) and Na2HPO3 buffer (pH 11−12). The pH stability test was performed by pre-incubating the recombinant lipase KV1 in various buffers (pH 4−12) for 30 min at 200 rpm, and the stability was monitored for 24 h [Citation22].
Results and discussion
Cloning of lipase KV1 gene
Characterization of sequence similarity in distantly related proteins has been proven useful for understanding the evolution of gene families. Bacterial lipolytic enzymes are categorized based on the amino acid sequences and biological properties [Citation23]. Bacterial lipases are highly conserved within the sequences of the open reading frame (ORF); more variations can be observed above and below the ORF [Citation23]. Despite the limited information on bacterial lipases, cloning lipases derived from the same family has been shown possible [Citation23].
Cloning of the amplified lipase KV1 gene (954 bp) using the pGEM-T-easy vector (Promega, Madison, WI, USA) was facilitated by a single 3′-T overhang of the vector and a single 3′-A overhang of the PCR product. While the Taq DNA polymerase conferred a 3′-A overhang to the PCR product, the pGEM-T-easy vector supplied a linearized single 3′-T overhang to enable direct cloning of the PCR product. In this study, the primary screening of positive clones (lipase producers) on agar plates was carried out using tributyrin-LB agar (100 μg/mL ampicillin) (Supplementary Figure S1 (A,B)), the simplest triglyceride occurring in natural fats and oils. Tributyrin degradation by the microorganisms was evaluated by clear zones surrounding the lipolytic colonies, converting the fat to water-soluble butyric acid [Citation24,Citation25]. This method is preferred due to its sensitivity over other known detection methods [Citation13]. However, the hydrolysis of tributyrin may also be carried out by both lipases and esterases [Citation13]; hence a positive result in the tributyrin plate assay may not always indicative of true lipase activity. Therefore, additional complementary qualitative analyses using the more specific triolein-LB and rhodamine B-LB agar plates were performed to confirm the true lipase activity in the recombinant lipase KV1. It was observed that the positive clone formed colonies with intense blue colouration on the triolein-LB plates (Supplementary Figure S1 (C,D)), indicating the positive lipase activity and presence of the lipase KV1 gene insert. The blue colour formation on the colonies showing positive lipase activity was attributable to the reduction in pH from free oleic acid, resulting from the hydrolysis of triolelglycerol substrate by the recombinant lipase KV1 [Citation24]. A positive lipase activity on the rhodamine B-LB plate (Supplementary Figure S1 (E,F)) appeared as orange fluorescence due to excited dimers of rhodamine B and uranyl-fatty acid on the agar plate upon exposure to ultraviolet (UV) light (350 nm). This observation further affirmed the identity of lipase KV1 as a true lipase [Citation13,Citation25].
Construction of His-LipKV1 expression plasmid
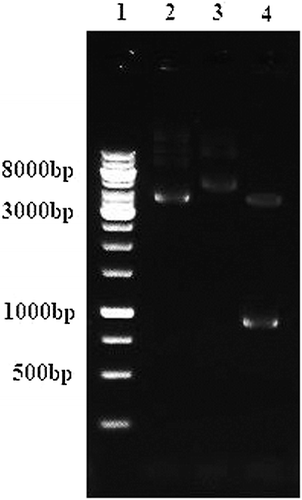
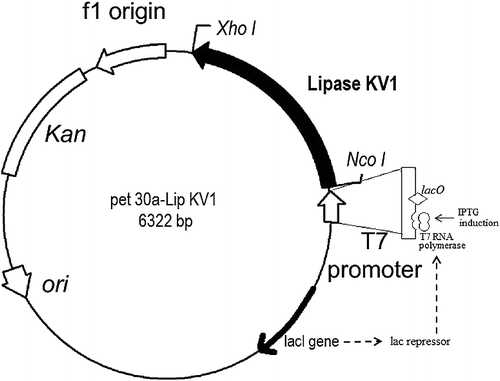
The purified lipase KV1 gene was inserted into the Nco1 and Xho1 sites of pET-30a expression vector. The resulting plasmid pet30a-lipKV1 encoded the recombinant HisTag-LipKV1 fusion protein with 6 × histidine-tag at the C-terminal. To identify the recombinant plasmid, restriction enzymes Nco1 and Xho1 were used and, fragments (5368 bp and a 954 bp) were characterized and separated on a 1% agarose gel (). The restriction enzymes digestion assay shows that the pet30a-lipKV1 expression plasmid was constructed successfully. Hence, the optimization for the recombinant lipase KV1 was carried out.
Figure 1. Analysis of recombinant plasmid pET30a/KV1 through double digestion. Lane 1: Marker 1 kb; Lane 2: empty plasmid pET30a (+) (5422 bp); Lane 3: recombinant plasmid pET30a/KV1 (6322 bp); Lane 4: recombinant plasmid pET30a/KV1 digested with Xho1 (5368bp) and Nco1 (954bp).
Optimization of recombinant lipase KV1 gene expression using RSM
ANOVA
A good combination of expression system and host is necessary to obtain high-level expression. E. coli remains a valuable organism for overproduction of intracellular recombinant proteins [Citation25], although not every gene can be expressed effectively in E. coli. A high-level gene expression is achieved by increasing the amount of expressed protein up to 50% of the total cell protein through gene manipulation, as well as proper selection of the vector and host combination before the recombinant plasmid of lipase KV1 can be transformed into E. coli BL21 (DE3) for protein expression. For the overexpression of the recombinant lipase KV1, physical and chemical factors, viz. OD600 before induction, IPTG concentration, induction temperature and time, were considered. The variables are relevant in the production and the correct folding of the protein overexpressed in E. coli [Citation26]. Therefore, the induction conditions were optimized for enhancing the expression of the recombinant lipase KV1.
The selected CCD model consisting of four factors, viz. (A) OD600; (B) IPTG concentration, (C) post-induction temperature and (D) post-induction time, was studied at five different levels (−2, −1, 0, +1, +2) (). The design and responses of the experimental conditions are presented in . The predicted values obtained with a model fitting technique were sufficiently correlated with the observed values, fitted well with various models (linear, two-factorial, quadratic and cubic). The ANOVA indicated that the activity of recombinant lipase KV1 was most suitably described with a quadratic polynomial model, represented by(2)
(2) where Y is the response (lipase activity), while A, B, C and D are the coded terms for the four variables, i.e. OD600 before induction, inducer (IPTG) concentration, post-induction temperature and post-induction time, respectively.
The expression of the recombinant lipase KV1 was the measured response in the experiments. Plus (+) and minus (−) symbols represent the positive and negative effect (EquationEquation (2)(2)
(2) ). The responses under different combinations were analysed using ANOVA which classified and cross-classified statistical results as well as tested for significant differences in the means of the specified classifications. The ANOVA results for the model of recombinant lipase KV1 production are presented in . The P-values were used to test the significance (P < 0.05) of the coefficients. outlines the regression coefficients estimated by the model for bi, bij and bii, along with the significance levels of the terms. It was apparent that each linear term (A, B, C and D) was significant at influencing the activity of the produced recombinant lipase KV1 protein. The regression analysis demonstrated the quadratic terms (A2, B2, C2 and D2) as well as the product terms (AD, AC, BD and CD) were statistically significant (P < 0.05) in the models. It was apparent that the model was categorically significant, as indicated by the insignificant lack of fit (P > 0.05), as well as a large F-value (100.26) and a very low probability value (< 0.0001). The values R2 = 0.9894 and adjusted R2 = 0.9796 were in reasonable agreement; with both the Pred R2 = 0.9486 and Adj R = 0.9796, respectively ().
Table 3. ANOVA and model coefficients.
Model diagnostics
The adequacy of the adopted model was verified using the normal (%) probability plot of the ‘Studentized’ residuals (). It is a major diagnostic tool which detects and accounts for the systematic departure from the assumptions that errors are normally distributed and independent of each other [Citation27]. The results revealed that the errors of the models for the production of the recombinant lipase KV1 showing high activity were normally distributed and insignificant ().
Figure 2. Normal (%) probability plot of the ‘Studentized’ residuals for the model of lipase KV1 production.
Optimization of the overexpression protocol
Effect of OD600 and post-induction temperature
It has been previously demonstrated in several studies that the four most relevant variables influencing recombinant protein expression are cell density before induction (OD600), inducer concentration, post-induction temperature and the duration of induction [Citation26–28]. The expression of recombinant lipase KV1 for the different levels of variables was predicted using surface and contour plots involving an infinitive number of combinations of two tested variables with the two remaining variables were held constant at their central values.
In general, the cultivation temperature and OD can have significant influence on the expression levels of proteins [Citation15]. Both process variables were among several determining factors identified to affect the overexpression of proteins, significantly altering the activity the recombinant lipase KV1. depicts the mutual effect of OD600 (A) and post-induction temperature (C) at influencing the activity of the expressed recombinant lipase KV1. The rather sharp curvature of the surface plot seen here ((A)) denotes that the surface was confined within the smallest ellipse in the contour plot ((B)). It was evident that the cell density of recombinant lipase KV1 before induction and the post-induction temperature may be critical factors for the expression of recombinant proteins. The highest activity of the recombinant lipase KV1 was as high as approximately 307 U/mL, when both the OD600 and post-induction temperature were close to their mid values of 0.6 nm and 40 °C, respectively. The ANOVA results indicated that the mutual interaction of these two variables was significant (P = 0.007). The linear term for the post-induction temperature (F-value = 14.44) was more significant than the OD600 (F-value = 5.65) at influencing the activity of the expressed recombinant lipase KV1 (). Increased activity of the recombinant lipase KV1 was observed with the increase of both cell density and post-induction temperature up to their central points, beyond which the activity started to decline (). Such an observation was consistent with the positive term in the model equation (+6.8AC), implying a synergistic effect between the two variables. Similar results have also been reported in previous studies [Citation29–31].
Figure 3. Graphs depicting the response surface plot (A) and contour plot (B) showing the effect of different temperature and OD600 as well as their mutual interaction on the activity of the recombinant lipase KV1.
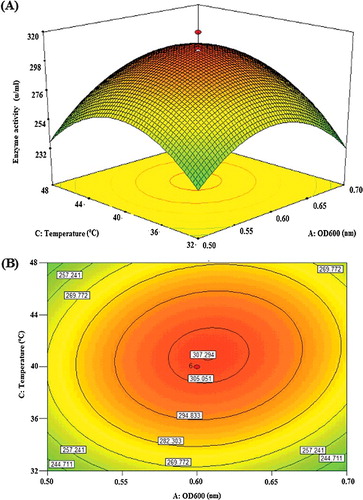
For the recombinant lipase KV1, a moderate post-induction temperature gave the highest lipase activity. This indicates that, at higher post-induction temperature, the overexpressed proteins may be disturbed by the presence inclusion bodies [Citation32]. At moderate induction temperature (40 °C), the strength of improper hydrophobic interactions was diminished, hence improving the activity of the recombinant lipase KV1. At that optimum temperature, too, the overexpression may probably induce the highly conserved heat shock response for synthesizing heat shock proteins. Such heat shock proteins as chaperones would assist in the folding of newly synthesized proteins, preventing heat-induced unfolding and irreversible aggregation of proteins [Citation33]. Therefore, the optimum post-induction temperature of 40 °C was used for the recombinant lipase KV1 evaluated here.
Cell density before induction can be a critical factor for the expression of recombinant proteins [Citation26]. The results showed that the induction of recombinant lipase KV1 IPTG at OD600 of 0.6 (i.e. mid-log phase) was optimum to yield the highest lipase activity, consistent with that reported by previous researchers [Citation26–28,Citation34]. Although induction is generally performed at early mid-log phase, studies have also reported on induction at the late-log phase or even stationary phase that yielded good results [Citation31]. In our study, cell density was measured at OD600 of 0.6, as at this point, the recombinant lipase KV1 cells reached their exponential phase for growth, including the expression of chaperones that would help to remove misfolded proteins. At this phase, the vast majority of recombinant lipase KV1 cells were alive and healthy, hence ideal for overexpression. Beyond the exponential phase, the recombinant lipase KV1 population was initially stabilized, prior to entering the death phase. This was consistent with indication made by previous studies that the maximum lipase production can be observed when induced at the mid-log phase of growth (OD600 of 0.6), and decreased rapidly after that [Citation35,Citation36].
Effect of OD600 and post-induction time
The overexpression of a recombinant enzyme can also be influenced by the post-induction time [Citation31]. The interactive effect of OD600 (A) and post-induction time (D) was examined and the results are illustrated in . For this assessment, the post-induction temperature and concentration of IPTG were kept constant at their central values of 40 °C and 0.5 mmol/L, respectively. The mutual interaction of both factors (AD) was highly significant (P = 0.0078) with a positive coefficient (+6.68AD) (EquationEquation (2)(2)
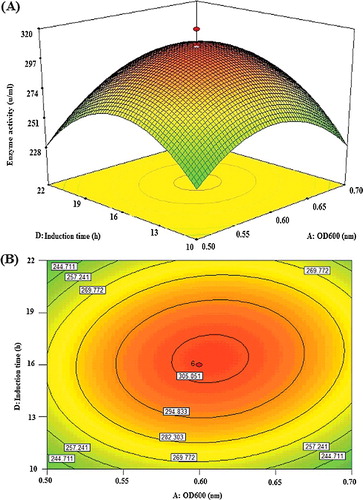
(2) ), indicating synergistic interaction favouring the overexpression of highly active recombinant lipase KV1. As indicated in , the effect of OD600 (F = 5.65) was marginally significant than that of post-induction time (F = 4.84). The highly elliptical response surface plot ((A)) clearly indicates the highest activity of the recombinant lipase KV1 (300.6 U/mL) when both variables, OD600 (A) and post-induction time (D) were close to their central values, OD600 of 0.6 and 16 h, respectively. The findings accentuated that post-induction time was an important factor to influence the expression level of intracellular recombinant lipase KV1. In the present study, the post-induction time of 16 h was found as optimal for affording acceptable expression of intracellular recombinant lipase KV1. It was a suitable duration to allow the overall correction folding of the lipase, as well as the accumulation of recombinant lipase KV1 in E. coli. Similar significant influence of post-induction time on the high expression of recombinant protein has also been reported previously [Citation26,Citation27,Citation35,Citation36]. Although longer post-induction time may enhance the expression level for recombinant lipase KV1, the approach may not be economical from the production perspective [Citation36]. Therefore, 16 h post-induction time was selected as one of the optimized culture conditions in this present study.
Figure 4. Graphs depicting the response surface plot (A) and contour plot (B) showing the effect of different induction time and OD600 and their mutual interaction in the lipase KV1 enzyme activity.
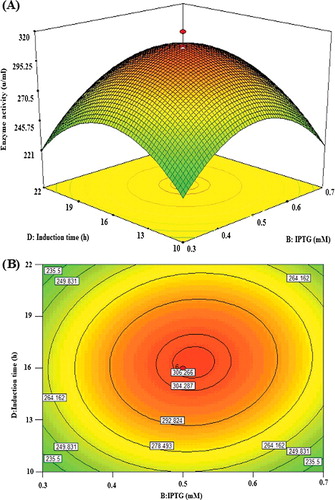
Effect of IPTG concentration and post-induction time
Considering that IPTG has high cost and is potentially toxic to cells, it is important to determine its optimum concentration for induction [Citation37]. The interactive effect of IPTG concentration (B) and post-induction time (D) was examined and the results are illustrated by both surface ((A)) and contour ((B)) plots. The ANOVA results () clearly revealed that the effect of IPTG concentration (F = 0.0056) was more significant than that of post-induction time (F = 4.84), and likewise, their mutual interaction was significant (P = 0.037). The expression of the recombinant lipase KV1 increased with time for up to 16 h as well as IPTG concentration up to its midpoint (0.5 mmol/L), reaching the peak of lipase activity at 300.6 U/mL (). Beyond this, the expression started to decline, showing that the addition of more than 0.5 mmol/L of IPTG did not improve the activity of the overexpressed recombinant lipase KV1.
Figure 5. Graphs depicting the response surface plot (A) and contour plot (B) showing the effect of different IPTG concentration and induction time, and their mutual interaction in the lipase KV1 enzyme activity.
In general, the IPTG concentration depends on the type of protein being expressed, and therefore this effect was examined here. It was observed that the optimum concentration of IPTG to induce the overexpression of the recombinant lipase KV1 was 0.5 mmol/L. Whereas a study [Citation38] indicated that IPTG concentrations of less than 1 mmol/L did not affect the specific growth rate or maximum cell concentration of E. coli, Nthangeni et al. [Citation19] reported that only 0.5 mmol/L of IPTG was needed to induce the expression of lipase from B. licheniformis using pET20b(+) and E. coli JM109 (DE3) as vector and host, respectively. The concentration (0.5 mmol/L) was found adequate to induce the lac operator regulated promoters to increase overexpression. This indicated the presence of lac operator in pET-30a that regulated the expression of lipase KV1, which was cloned downstream from the operator. The added 0.5 mmol/L of IPTG was sufficient to bind to the repressor to activate the expression of the lipase KV1 gene (). In this aspect, the present research further supports that the concentration of IPTG is an important factor for the expression of recombinant lipases, especially that of lipase KV1 in E. coli.
Figure 6. Map of the pET30a(+) – Lipase KV1 expression vector. Note: The lipase KV1 gene was cloned into the pET30a(+) vector between NcoI and XhoI restriction sites. This vector allows C-terminal fusion of recombinant protein with a His6 tag and contains a kanamycin resistant gene under the control of a T7 promoter.
Effect of post-induction time and post-induction temperature
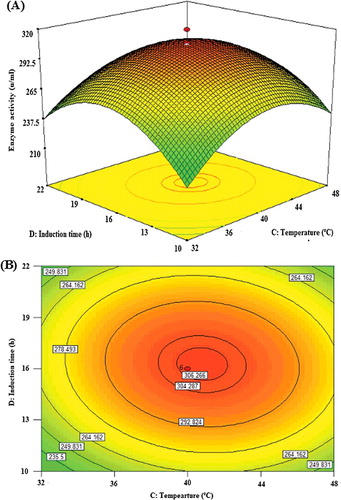
The post-induction time and the post-induction temperature are two key factors affecting protein expression [Citation36]. illustrates the interaction of post-induction time and post-induction temperature when OD600 and IPTG concentration were at their central values as shown in both the surface ((A)) and contour ((B)) plots. As indicated by the linear coefficients and F-value (), the effect of the post-induction temperature (14.44) was more highly significant than that of post-induction time (4.84). Their mutual interaction was found to be significant (P = 0.036). Decreasing the post-induction temperature down to a certain point as well as increasing the post-induction time within the experimental limit could increase lipase KV1 expression. The best post-induction time and temperature for protein expression were determined at 16 h and 40 °C, respectively. The interaction of the variables post-induction time and temperature was significant and showed a negative impact. The negative effect of the two variables was verified by the negative sign of the coefficient CD in EquationEq. (2)(2)
(2) . This is probably due to the lowering of the post-induction temperature that caused reduction in the rate of protein synthesis, and subsequently formation of inclusion bodies. On the other hand, the high temperature (56 °C) would promote growth with high lipase KV1 expression, despite being detrimental to the expression of the recombinant lipase KV1 protein. High growth rates tend to elevate the probability of losing the pET-30a plasmid, as well as stimulating mispartition of E. coli in several other studies [Citation13,Citation26].
Figure 7. Graphs depicting the response surface plot (A) and contour plot (B) showing the effect of induction time and temperature, and their mutual interaction in the lipase KV1 enzyme activity.
Validation of the model
The optimization of recombinant lipase KV1 production was performed using the “point optimization” technique of the Design Expert 7.1.6. Experiments (in triplicates) yielded the maximum recombinant lipase production of 320.9 U/mL, in good agreement with the predicted 321.7 U/mL. The optimum experimental conditions were obtained at OD600 of 0.6, 0.52 mmol/L IPTG, 40 °C post-induction temperature and 16 h post-induction time. Since the predicted and experimental values were highly similar, the adequacy and validity of the CCD model were fully justified ().
Table 4. Validation of model showing the production of recombinant lipase KV1 at optimum level of all variables.
Purification of recombinant lipase KV1
The choice of a suitable purification method that retains the structural integrity and biological activity of the recombinant proteins of interest, while avoiding contamination, is widely acknowledged as a significant challenge in biotechnology [Citation39]. By using such technique, lipase KV1 can be isolated based on the reaction of electron donor groups on the protein surface, for example, histidine in this study, with electron receptors like Ni2+ immobilized within an inert phase [Citation40]. The protein of interest was then bound to the nickel ion-charged NTA resin, followed by washing using an elution buffer. In this study, immobilized-metal (Ni-NTA agarose) affinity chromatography proved successful in the purification of the protein of interest, lipase KV1, with a relatively high homogeneity ().
Figure 8. SDS-PAGE (A) and Western blotting (B) analyses showing the purification of recombinant lipase KV1 expressed in E. coli BL21 (DE3). (A) Proteins recovered during the various purification steps were analysed by SDS-PAGE. Lane M: prestained standard protein molecular weight markers; Lane 1: lysate supernatants of uninduced transformant harbouring empty pET-30a(+); Lane 2: lysate supernatants of induced transformant harbouring empty pET-30a(+); Lane 3: uninduced intracellular lysate supernatants pET-lipKV1; Lane 4: induced intracellular lysate supernatants pET-LipKV1 and Lane 5: purified fraction from Ni–NTA column eluted by buffers containing 200 mmol/L imidazole. (B) Western blot analysis of purification steps probed with an anti-His antibody. The samples are the same as those described for panel (A).
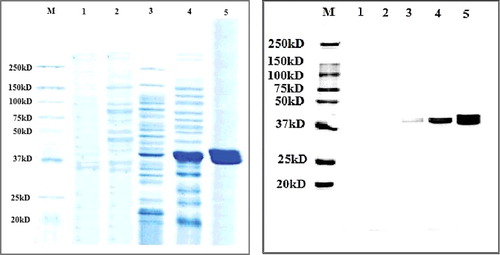
In this study, the T7-based pET vector for the E. coli expression system was chosen to produce the recombinant KV1 protein, as this expression system has been reported to be fast growing, as well as producing high yield of the target protein. In addition to being compatible with the cloned gene, this vector is recognized for its acceptable conditions of expression and efficiency, and most importantly, for the fact that it facilitates purification due to presence of the His6-tag sequence [Citation41]. The recombinant lipase KV1 was then purified to homogeneity by His-Bind resin affinity chromatography with a six-histidine tag at the C-terminus. SDS-PAGE ((A)) and Western blot ((B)) analyses using anti-HIS antibody showed a distinctly expressed protein band of 39 kDa. The obtained protein mass was in near agreement with the mass of the wild-type lipase KV1 (37 kDa). The additional 20 amino acid residues in the recombinant lipase KV1 corresponded to the C-terminus 6 × histidine-tag and the linker region. Analysis of the specific activity of the purified recombinant and wild-type lipase KV1 was executed using a standardized protein concentration of 1.2 mg/mL. The specific activity of the purified recombinant lipase KV1 was 233.4 U/mg (), 7.0-fold higher than that of the purified wild-type lipase (33.5 U/mg) (data not shown). Noteworthy, this 7.0-fold increase in the specific activity of the purified recombinant lipase compared with the wild-type lipase KV1, was coupled with higher gene expression of recombinant lipase KV1 than that reported for the thermostable lipases from Bacillus thermoleovorans ID-1 [Citation42] and Geobacillus sp. strain T1 [Citation25]. Although the purified wild-type lipase KV1 has great commercial significance, its utilization may be limited due to the low yields of the produced enzyme. In this context, the use of the strategy for production of recombinant KV1 lipase described here may overcome such constraints, while facilitating large-scale production of the lipase useful for various industrial applications.
Table 5. Purification of the recombinant lipase KV1 from A. haemolyticus.
The findings of the SDS-PAGE and western blotting analyses of the different fractions obtained from the protein purification step indicated that the His-6 sequence of the target recombinant lipase KV1 protein had effectively and completely bound to the column, with each fraction (fractions 3–5) containing highly pure protein (). These findings were consistent with previous studies [Citation6] conducted on the optimization of the expression and purification of recombinant proteins. The His6-tag sequence aids the selective binding of the expressed protein to the nickel column without considerable impact on protein structure [Citation40]. Therefore, the results from the present study agree with the concept that E. coli BL21 (DE3) as a host could be a function of the pET30a vector [Citation41]. This observation is in agreement with earlier reports, showing the significant effect of E. coli BL21 (DE3) and pET30a on the expression of recombinant proteins [Citation35,Citation36].
Characterization of the purified recombinant lipase KV1
Effect of temperature on lipase activity and stability
The effect of temperature was evaluated in this study and the relevant data are presented in . The activity profile for the recombinant lipase KV1 was determined from 5 to 80 °C. The results showed that 40 °C (100% relative activity) was the optimum temperature to give the highest lipase activity (305.5 U/mL). The enzyme activity dropped when the reaction temperature was beyond 50 °C ((A)). The purified recombinant lipase KV1 exhibited a broad temperature profile, similar to that of the wild-type lipase KV1 (data not shown). It was clear that cloning of the lipase KV1 gene into E. coli did not change the optimal temperature of the recombinant lipase KV1, but favourably enhanced the activity of lipase. The activity of the recombinant lipase KV1 was seen to improve with the increase in reaction temperature probably due to the enhanced effective collisions between the lipase and substrate molecules following the elevated overall kinetic energy within the system [Citation10]. The highest activity seen in the recombinant lipase KV1 at 40 °C agrees well with previous reports on greater unfolding of protein structure [Citation43], reducing lipases rigidity [Citation43], while catalysing more competently. Recombinant lipase KV1 exhibited a broad temperature profile, whereby the lipase remained active and retained its half-life (>50%) even at temperatures ranging from 40–70 °C ().
Figure 9. Effect of temperature on the activity (A) and stability (B) of the purified recombinant lipase KV1. Note: Relative activities are the average values of triplicate measurements.
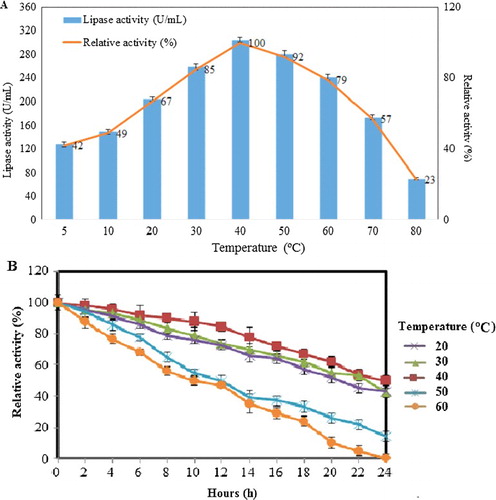
The relative activity of recombinant lipase KV1 was the lowest (128.1 U/mL, 42%) at 5 °C due to the lipase KV1 structure being too rigid and unable to unravel into its catalytically active form [Citation10]. Similarly, low activity of lipase KV1 (70.2 U/mL, 23%) was observed at 80 °C ((A)), attributable to the breaking of essential intramolecular bonds [Citation10], which causes excessive unfolding of the lipase KV1 protein [Citation44] and the subsequent irreversible thermal inactivation of the lipase. Although increasing the temperature of the systems would initially result in more molecules reaching the activation energy, elevating the assay temperature further would increase the internal energy of the lipase KV1 molecules, which causes the weak hydrogen and/or van der Waals bonds that hold the three-dimensional shape of the active lipase KV1 protein to break, hence denaturing the lipase in the process. Moreover, the elevated temperature of reaction may cause over-flexibility of the active-site of recombinant lipase KV1, leading to gradual distortion of its structure. Such conditions would render the recombinant lipase KV1 inactive along with the concomitant drop in its catalytic activity. Our observation corroborates previous reports by similar studies [Citation10,Citation44].
Assessment of the enzyme stability showed that the recombinant lipase KV1 retained its structural stability and was catalytically competent over a wide range of temperatures for a long time (up to 24 h) ((B)). The recombinant lipase KV1 retained over 85% of its relative activity between 20 °C and 40 °C after 12 h of incubation. At the optimum temperature (40 ˚C), the half-life of the lipase was an impressive 24 h, characteristically similar to the wild-type lipase KV1. Hence, it is affirmed that the cloning and the overexpression of lipase KV1 had been successful in retaining the exceptional qualities of the wild-type KV1 in the recombinant lipase.
Effect of pH on lipase activity and stability
It would be an interesting factor to observe whether cloning of the lipase KV1 gene into a vector would alter the optimal pH of lipase KV1. The lipase assay was conducted at various pH levels for 30 min. The results revealed that the purified recombinant lipase KV1 retained its activity over a broad pH range (7–12) with the optimum pH being 8 (100% relative activity), similar to that of wild-type lipase ((A)). The activity of the recombinant lipase KV1 of less than 60% was achieved at acidic conditions of pH 4 (112.9 U/mL, 38%), pH 5 (118.9 U/mL, 40%) and pH 6 (169.4 U/mL, 57%) ((A)). This implies that the recombinant lipase KV1 requires the proper ionic form to maintain its active site conformation for correct binding with the substrate; deactivating the structure and activity of recombinant lipase KV1 at lower pH range. However, the activity of lipase KV1 remained high at alkaline conditions (pH 8−12), retaining approximately 70% of its relative activity up to pH 12. This strongly implies that the recombinant lipase KV1 demonstrated alkaline-stable properties, like its wild-type counterpart. The remarkably wide pH range of the recombinant lipase KV1 suggests its possible wide utilization for various commercial applications.
Figure 10. Effect of pH on the activity (A) and stability (B) of the purified recombinant lipase KV1. Note: Relative activities are the average values of triplicate measurements.
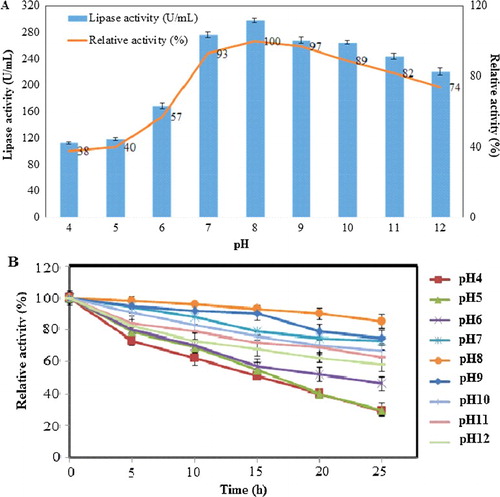
The present study also carried out time-course profiling to assess the effects of various pH on the stability of the recombinant lipase. The results showed that the recombinant lipase KV1 was stable in the upper pH range. At the alkaline pH range (pH 8−12), relative activities of >90% and >70% were obtained for lipase KV1 incubated for 5 and 24 h, respectively ((B)). The exceptionally lengthy half-life of the recombinant lipase KV1 (up to 24 h in buffers pH 8−12) was substantially longer that those described in literature [Citation12,Citation23,Citation25,Citation45]. Earlier reports associated the exceptional pH stability of certain enzymes, especially alkaline-stable or alkalophilic enzymes, with the presence of a higher number of acidic surface amino acids (negatively charged) [Citation46] on the protein. Such adaptation is, therefore, expected for the KV1 lipase, as this would justify the highly active and stable lipase activity at the higher pH range. Theoretically, the surplus in acidic amino acids would beneficially prepare the lipase to better accommodate structural changes in the protein of the enzyme associated with pH-induced local folding events. Hence, this assumption can be further clarified with in silico works involving homology modelling, molecular dynamics simulations and substrate docking studies.
Conclusions
In this study, optimization for the overexpression of the recombinant KV1 lipase using CCD was successfully carried out. Under the optimized induction conditions of OD600 before induction of 0.6, IPTG concentration of 0.5 mmol/L, post-induction temperature of 40 °C and post-induction time of 16 h, the specific activity of the recombinant lipase KV1 was improved by 70% over that of the wild-type lipase. The recombinant lipase KV1 showed optimum temperature and pH at 40 °C and pH 8.0, respectively. Pertinently, the remarkable stability of the KV1 lipase over a broad range of pH values (pH 7−12), as well as an optimum activity at 40 °C suggests that the recombinant lipase KV1 may be suitable for a wide range of industrial applications, e.g. as an active component in the detergent industry, a flavour enhancer in the food industry, fat and pitch remover in the paper and pulp industry, environmental bioremediation and molecular biology. Alkaline-stable enzymes such as lipase KV1 are, therefore, useful in biotechnology-based industries in order to shorten the production time, minimize the energy consumption and prevent undesired chemical transformations, as well as the loss of volatile compounds. As enzyme technology is a cleaner and greener technology in comparison to the chemical-based technology, the uses of lipases will prospectively be useful in commercial manufacturing activities.
Supplemental_Data.pdf
Download PDF (433.5 KB)Acknowledgments
The authors acknowledge the valuable help and suggestions provided by their colleagues.
Additional information
Funding
Related Research Data
References
- Jayaprakash A, Ebenezer P. Optimization of Aspergillus japonicus lipase production by response surface methodology. J Acad Indust Res. 2012;1(1):23–30.
- Mariod AA, Salaheldeen M. Oilseed crops and biodiesel production: present and future prospects. In: Ahmad P, editor. Oilseed crops: yield and adaptations under environmental stress. Chichester (UK): Wiley-Blackwell; 2017. p. 52–79.
- Sharma R, Thakur V, Sharma M, et al. Biocatalysis through thermostable lipases: adding flavor to chemistry. In: Satyanarayana T, Littlechild J, Kawarabayasi Y, editors. Thermophilic microbes in environmental and industrial biotechnology. Dodrecht (The Netherlands): Springer; 2013. p. 905–927.
- Snellman EA, Colwell RR. Acinetobacter lipases: molecular biology, biochemical properties and biotechnological potential. J Ind Microbiol Biotechnol. 2004;31(9):391–400.
- Palomares LA, Estrada-Moncada S, Ramírez OT. Production of recombinant proteins: challenges and solutions. In: Balbás P, Lorence A, editors. Recombinant gene expression: reviews and protocols. 2nd ed. Totowa (NJ): Humana Press; 2004. p. 15–51. ( Methods in Molecular Biology, Vol. 267).
- Rosano GL, Ceccarelli EA. Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol. 2014 [cited 2017 Jul 02];5:172. DOI: 10.3389/fmicb.2014.00172
- Zhao X, Li G, Liang S. Several affinity tags commonly used in chromatographic purification. J Anal Meth Chem. 2013 [cited 2017 Jul 02];2013:581093; [8 p.] DOI: 10.1155/2013/581093
- Ferrer-Miralles N, Saccardo P, Corchero JL, et al. General introduction: recombinant protein production and purification of insoluble proteins. In: García-Fruitós E, editor. Insoluble proteins: methods and protocols. New York (NY): Humana Press; 2015. p. 1–24. ( Methods in Molecular Biology, vol. 1258)
- Marzuki NH, Huyop F, Aboul-Enein HY, et al. Modelling and optimization of Candida rugosa nanobioconjugates catalysed synthesis of methyl oleate by response surface methodology. Biotechnol Biotechnol Equip. 2015;29(6):1113–1127.
- Wahab RA, Basri M, Rahman RN, et al. Enzymatic production of a solvent-free menthyl butyrate via response surface methodology catalyzed by a novel thermostable lipase from Geobacillus zalihae. Biotechnol Biotechnol Equip. 2014;28(6):1065–1072.
- Chauhan M, Chauhan RS, Garlapati VK. Modelling and optimization studies on a novel lipase production by Staphylococcus arlettae through submerged fermentation. Enzyme Res. 2013 [cited 2017 Jul 02];2013:353954; [ 8 p.]. DOI: 10.1155/2013/353954
- Farrokh P, Yakhchali B, Asghar Karkhane A. Cloning and characterization of newly isolated lipase from Enterobacter sp. Bn12. Braz J Microbiol. 2014;45(2):677–687.
- Kouker G, Jaeger KE. Specific and sensitive plate assay for bacterial lipases. Appl Environ Microbiol. 1987;53(1):211–213.
- Gabor EM, de Vries EJ, Janssen DB. Efficient recovery of environmental DNA for expression cloning by indirect extraction methods. FEMS Microbiol Ecol. 2003;44(2):153–163.
- He YQ, Tan TW. Use of response surface methodology to optimize culture medium for production of lipase with Candida sp. 99-125. J Mol Catal B Enzym. 2006;43(1):9–14.
- Gunst RF. Response surface methodology: process and product optimization using designed experiments. Raymond H. Myers and Douglas C. Montgomery [book review]. Technometrics. 1996;38(3):284–286.
- Benoit I, Coutard B, Oubelaid R, et al. Expression in Escherichia coli, refolding and crystallization of Aspergillus niger feruloyl esterase A using a serial factorial approach. Prot Express Purif. 2007;55(1):166–174.
- Kwon DY, Rhee JS. A simple and rapid colorimetric method for determination of free fatty acids for lipase assay. J Am Oil Chem Soc. 1986;63(1):89–92.
- Nthangeni MB, Patterton HG, van Tonder A, et al. Over-expression and properties of a purified recombinant Bacillus licheniformis lipase: a comparative report on Bacillus lipases. Enzyme Microb Technol. 2001;28(7):705–712.
- Brault G, Shareck F, Hurtubise Y, et al. Isolation and characterization of EstC, a new cold-active esterase from Streptomyces coelicolor A3 (2). PloS One. 2012 [cited 2017 Jul 02];7(3):e32041. DOI: 10.1371/journal.pone.0032041.
- Zheng X, Chu X, Zhang W, et al. A novel cold-adapted lipase from Acinetobacter sp. XMZ-26: gene cloning and characterization. Appl Microbiol Biotechnol. 2011;90(3):971–980.
- Kasana RC, Kaur B, Yadav SK. Isolation and identification of a psychrotrophic Acinetobacter sp. CR9 and characterization of its alkaline lipase. J Basic Microbiol. 2008;48(3):207–212.
- Asoodeh A, Emtenani S, Emtenani S. Expression and biochemical characterization of a thermophilic organic solvent-tolerant lipase from Bacillus sp. DR90. Protein J. 2014;33(5):410–421.
- Lanka S, Pydipalli M, Latha JN. Optimization of process variables for extracellular lipase production from Emericella nidulans NFCCI 3643 isolated from Palm Oil Mill Effluent (POME) dump sites using OFAT method. Res J Microbiol. 2015;10(2):38–53.
- Leow TC, Rahman RN, Basri M, et al. A thermoalkaliphilic lipase of Geobacillus sp. T1. Extremophiles. 2007;11(3):527–535.
- Papaneophytou CP, Rinotas V, Douni E, et al. A statistical approach for optimization of RANKL overexpression in Escherichia coli: purification and characterization of the protein. Protein Expr Purif. 2013;90(1):9–19.
- Papaneophytou CP, Kontopidis GA. Optimization of TNF-α overexpression in Escherichia coli using response surface methodology: purification of the protein and oligomerization studies. Protein Expr Purif. 2012;86(1):35–44.
- Muntari B, Amid A, Mel M, et al. Recombinant bromelain production in Escherichia coli: process optimization in shake flask culture by response surface methodology. AMB Express. 2012 [cited 2017 Jul 02];2(1):12; [9 p.]. DOI:10.1186/2191-0855-2-12
- Volontè F, Piubelli L, Pollegioni L. Optimizing HIV-1 protease production in Escherichia coli as fusion protein. Microb Cell Fact. 2011;10(1):53; [10 p.]. DOI: 10.1186/1475-2859-10-53
- Larentis AL, Argondizzo AP, dos Santos Esteves G, et al. Cloning and optimization of induction conditions for mature PsaA (pneumococcal surface adhesion A) expression in Escherichia coli and recombinant protein stability during long-term storage. Protein Expr Purif. 2011;78(1):38–47.
- Akbari V, Sadeghi HM, Jafarian-Dehkordi A, et al. Optimization of a single-chain antibody fragment overexpression in Escherichia coli using response surface methodology. Res Pharm Sci. 2015;10(1):75–83.
- Upadhyay V, Singh A, Panda AK. Purification of recombinant ovalbumin from inclusion bodies of Escherichia coli. Protein Expr Purif. 2016;31(117):52–58.
- Hendrick JP, Hartl FU. Molecular chaperone functions of heat-shock proteins. Annu Rev Biochem. 1993;62(1):349–384.
- Gupta N, Sahai V, Gupta R. Alkaline lipase from a novel strain Burkholderia multivorans: statistical medium optimization and production in a bioreactor. Process Biochem. 2007;42(4):518–526.
- Pournejati R, Karbalaei-Heidari HR, Budisa N. Secretion of recombinant archeal lipase mediated by SVP2 signal peptide in Escherichia coli and its optimization by response surface methodology. Protein Expr Purif. 2014;30(101):84–90.
- Pan H, Xie Z, Bao W, et al. Optimization of culture conditions to enhance cis-epoxysuccinate hydrolase production in Escherichia coli by response surface methodology. Biochem Eng J. 2008;42(2):133–138.
- Bernaudat F, Frelet-Barrand A, Pochon N, et al. Heterologous expression of membrane proteins: choosing the appropriate host. PloS one. 2011;6(12):e29191; [17 p.]. DOI:10.1371/journal.pone.0029191
- Ramirez OT, Zamora R, Espinosa G, et al. Kinetic study of penicillin acylase production by recombinant E. coli in batch cultures. Process Biochem. 1994;29(3):197–206.
- Van Oss CJ. A review of: “Protein Purification, (Principles and Practice), RK Scopes, Springer-Verlag, New York, 1982; hardbound, 282 pages, $29.95” [book review]. Prep Biochem. 1984;14(1):89–90.
- Ley C, Holtmann D, Mangold KM, et al. Immobilization of histidine-tagged proteins on electrodes. Colloids Surf. 2011;88(2):539–551.
- Heidari R, Rabiee-Faradonbeh M, Darban-Sarokhalil D, et al. Expression and purification of the recombinant cytochrome P450 CYP141 protein of Mycobacterium tuberculosis as a diagnostic tool and vaccine production. Iran Red Crescent Med J. 2015 [cited 2017 Nov 16];17(6):e23191; [6 p.]. DOI: 10.5812/ircmj.23191v2
- Cho AR, Yoo SK, Kim EJ. Cloning, sequencing and expression in Escherichia coli of a thermophilic lipase from Bacillus thermoleovorans ID-1. FEMS Microbiol Lett. 2000;186(2):235–238.
- Daniel RM, Peterson ME, Danson MJ, et al. The molecular basis of the effect of temperature on enzyme activity. Biochem J. 2010;425(2):353–360.
- Kok RG, Thor JJ, Nugteren‐Roodzant IM, et al. Characterization of the extracellular lipase, LipA, of Acinetobacter calcoaceticus BD413 and sequence analysis of the cloned structural gene. Mol Microbiol. 1995;15(5):803–818.
- Sharma M, Chadha BS, Saini HS. Purification and characterization of two thermostable xylanases from Malbranchea flava active under alkaline conditions. Bioresour Technol. 2010;101(22):8834–8842.
- Bora L, Bora M. Optimization of extracellular thermophilic highly alkaline lipase from thermophilic Bacillus sp isolated from Hotspring of Arunachal Pradesh, India. Braz J Microbiol. 2012;43(1):30–42.