
ABSTRACT
Rhizobium sp. RC1 grows on haloalkanoic acid (haloacid) pollutants and expresses a haloacid permease (DehrP), which mediates the uptake of haloacids into the cells. For the first time, we report the homology model and docking analysis of DehrP and propose its putative binding residues. Ligand structures were retrieved from the ChemSpider database. The three-dimensional (3D) structure of DehrP was modelled based on the structure of Staphylococcus epidermidis glucose:H+ symporter (GlcPse) by Phyre2, refined by 3Drefine and evaluated by ProSA z-score, ERRAT and RAMPAGE. The 3D structure of the DehrP protein has 12 transmembrane helices. The overall quality factor of the model is ∼91%, with 93.6% of the residues in the favoured region and the z-score (−2.86) falls within the range (≤10) for a good model. Subsequent docking of monobromoacetate, monochloroacetate, dibromoacetate, dichloroacetate, trichloroacetate and 2,2-dichloropropionate ligands via AutoDock Vina1.1.2 showed that residues Gln133, Asp36 and Arg130 are the putative H+-binding site, while the probable haloacid interacting residues are Glu33, Trp34, Phe37, Phe38, Gln165 and Glu370. The DehrP-haloacid complexes exhibited binding affinities between −2.9 and −4.0 kcal/mol. Both the putative H+ and haloacid-binding sites of DehrP possibly aided in co-transportation of substrates H+ and haloacids into the bacterial cells through the alternating access mechanism, which occurs by formation of halogen bonds and van der Waals interactions with the substrates. Hence, site-directed mutagenesis on the DehrP binding residues could improve the haloacid-binding affinity for efficient haloacid degradation.
Introduction
The microbial metabolism of haloalkanoic acid (haloacid) pollutants depends on haloacid transport [Citation1,Citation2] and dehalogenation by the micro-organisms [Citation3,Citation4]. Some micro-organisms are unable to degrade haloacids [Citation5,Citation6] because the metabolism of these compounds does not support cell growth [Citation7]. Likewise, cases of haloacid selectivity by cells [Citation8–10] and haloacid cytotoxicity [Citation11,Citation12] have been reported. It is understood that dehalogenase-associated inducible haloacid transport proteins are involved in the uptake of haloacids into the cytoplasm of bacteria [Citation13–15]. Kinetics studies on Burkholderia caribensis MBA4 haloacid transporter (Dehp2) have shown that the protein prefers chloropropionate over monochloroacetate [Citation16]. Therefore, to carry out an efficient haloacid biodegradation process, it requires a firm understanding of the structure and mechanism of the haloacid transport proteins. So far, the three-dimensional (3-D) structure of haloacid transport proteins and their mechanism of action remain unknown.
Rhizobium sp. RC1 utilizes haloacids as the carbon and energy source by producing three distinct dehalogenases (D-2-haloacid dehalogenase; DehD, L-2-haloacid dehalogenase; DehL and dual isomeric haloacid dehalogenase; DehE) to cleave the carbon-halogen (C…X) bonds [Citation17–19]. Rhizobium sp. RC1 haloacid permease gene dehrP is located 511 bases upstream of the D-2-haloacid dehalogenase gene dehD, and encodes the haloacid transport protein (DehrP) [Citation15]. DehrP has the Major Facilitator Superfamily (MFS) transport protein conserved domain that is comprised of a sugar signature [Citation15] and is closely related to the Metabolite:H+ Symporter (MHS) family of haloacid transporters [Citation20] (Deh4p and Dehp2) from Burkholderia caribensis MBA4, a subfamily of the MFS [Citation21,Citation22].
Proteins from the six subfamilies of the MFS show low sequence similarities with unique substrate specificities, and distinct transport-coupling mechanisms. All of them share a common structural motif, known as the MFS fold [Citation23]. Therefore, these transporters conform to the same 3D structural arrangement, which consists of 12 transmembrane helices (TMs) arranged into two folded domains (N and C), in which each domain has six TMs [Citation24–31].
In this study, for the first time, we report the homology modelling and haloacid-binding analysis of Rhizobium sp. RC1 haloacid permease (DehrP). The 3D structure of DehrP was modelled based on the crystal structure of Staphylococcus epidermidis glucose:H+ symporter (GlcPse) [Citation32]. We conducted a comparative binding analysis of the putative haloacid-binding site of DehrP with six haloacid ligands (monobromoacetate, MBA; monochloroacetate, MCA; dibromoacetate DBA; dichloroacetate, DCA; trichloroacetate, TCA; and 2,2-dichroropropionate, 2,2-DCP) to identify the putative residues involved in haloacid transport. In this work, we also proposed the mechanism of haloacid transport carried out by DehrP.
Materials and methods
Sequence retrieval and alignment
The amino acid sequence of Rhizobium sp. RC1 haloacid permease (DehrP) [Citation15] was downloaded from the UniProtKB Database [Citation33] with accession number Q1M2W6. NCBI-Blastp [Citation34], PSI-Blast [Citation35] and UniProt-Blastp [Citation33] were used to analyse the amino acid sequence of DehrP for sequence homology. The DehrP sequence was aligned with S. epidermidis glucose:H+ symporter (GlcPse, Uniprot number: Q5HKL0) [Citation32] using Clustal Omega [Citation36].
Homology modelling and model evaluation
The 3D structure of DehrP was modelled based on the crystal structure of S. epidermidis glucose:H+ symporter (GlcPse, PDB accession number: 4LDS B) using the Phyre2 server [Citation37]. The 3D structure of DehrP (PDB file) was then submitted to 3Drefine server to optimize the hydrogen-binding network and to apply atomic-level energy minimization on the optimized model using physics and knowledge-based force fields [Citation38]. ProSA-web was used to calculate the z-score of the 3-D structure from a score and energy plot in order to check for potential errors in the model using its atomic coordinates [Citation39]. RAMPAGE server [Citation40] was used to generate a Ramachandran 2-D contour plot between Ψ (psi) and Φ (phi) torsion angles of each amino acid residue, to predict the stereochemical quality of the 3-D structure. Similarly, ERRAT (https://www.Shannon.mbi.ucla. edu/DOE/services/SV/) [Citation41] was used to evaluate the overall quality of the protein structure. The software uses an error function that is based on the statistics of non-bonded atom–atom interactions in the modelled structure in comparison to a database of reliable high-resolution structures. The DehrP 3D structure was then superimposed with the GlcPse (template) 3-D structure by the Chimera 1.11.2rc software [Citation42]. Based on the longer length of the GlcPse structure (425 residues), the stretch of residues from the C-terminus of Agrobacterium sp. NHG3 (with 98% sequence identity, see supplementary Figure S1) without corresponding residues in DehrP was added to the DehrP C-terminus before modelling. This is a technique typically adopted when an indirect threading approach to model proteins of unequal sequence length with the template of interest is used [Citation43].
Molecular docking
Ligand and receptor preparation
The 3D structures of D-glucose (DGlc), monobromoacetate (MBA), monochloroacetate (MCA), dichloroacetate (DCA), dibromoacetate (DBA), trichloroacetate (TCA) and 2,2-dichroropropionate (2,2-DCP) were downloaded from the ChemSpider [Citation44] ligand database under the following ChemSpider IDs: 96749, 5991, 452706, 24195, 5602522, 106518 and 2815701, respectively. The ligands were used as input files for AutoDock Tools 1.5.6 [Citation45]. The ligand rigid roots were automatically set and all possible rotatable bonds and torsions were defined as active. The docking simulation used the modelled DehrP 3-D structure (receptor) which consists of one single chain B (419 residues). DehrP in PDB format was submitted to the AutoDock Tools 1.5.6 [Citation45], after which polar hydrogen atoms were added and Kollman charges, atomic solvation parameters and fragmental volumes were assigned to the protein. After that, polar hydrogen atoms were added and Kollman charges, atomic solvation parameters and fragmental volumes were assigned to the protein. Chain B (425 residues) of the 3D structure of S. epidermidis glucose transporter (GlcPse, PDB: 4LDS) downloaded from PDB was also prepared as the control to be used in the docking procedure.
Docking procedure
Docking simulation was performed using AutoDock Vina 1.1.2 [Citation46], in which hydrogens and Kollman charges were assigned to the DehrP receptor protein, followed by conversion of the PDB file to pdbqt. Ligands were assigned with Gasteiger charges and non-polar hydrogens. Docking simulations were run using the Lamarckian Genetic Algorithm (LGA), which is known to be the most efficient and reliable method of AutoDock. The ligand cantered maps were generated by the AutoGrid program with a spacing of 0.200 Å and dimensions of 100 × 100 × 100 points. The grid box centre was set to coordinates 3.01841, 48.2993 and 67.4807 in x, y and z, respectively. The default settings were used for all other parameters. For each docking simulation, nine different conformers were generated, for which Chimera 1.11.2 software [Citation42] was used to visualize the obtained docking conformations. The AutoDock Vina 1.1.2 [Citation46] binding free energies (ΔGb in kcal/mol) for the seven ligand–receptor complexes for DehrP and GlcPse were then compared. The results were visualized in UCSF Chimera v.1.11.2rc [Citation42] and Discovery Studio v.16.1.0 [Citation47]. The different DehrP-haloacid binding conformations generated from this assessment were then used to propose the haloacid binding site and mechanism of haloacid transport.
Results and discussion
Sequence alignment
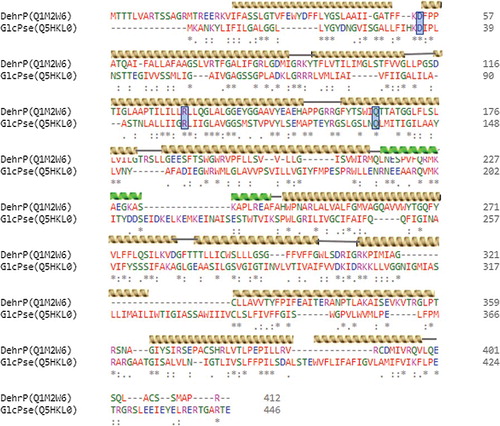
Comparative analysis of protein sequences by sequence alignment provides useful information during structural and functional analysis by revealing sequence–structure–function relationships [Citation48,Citation49]. PSI-Blast of the Rhizobium sp. RC1 haloacid permease sequence in the NCBI database affirmed that DehrP has 30% sequence identity with S. epidermidis glucose:H+ symporter (GlcPse, NCBI accession number: WP_002486092.1). An alignment of the two sequences revealed that DehrP and GlcPse share some common secondary structural elements (helices) and amino acids at specific locations (), including some glucose and H+-binding site residues.
Figure 1. Alignment of DehrP sequence with glucose symporter (GlcPse) template.
Homology modelling
As reported by Abramson et al. [Citation23], transporter proteins that belong to the MFS conform to the same 3D structure. Since DehrP is a member of the MFS [Citation15,Citation22], the protein is expected to follow a similar structure to the experimentally determined 3D structures of microbial MFS transporters. The DehrP sequence was threaded onto the crystal structure (at 3.2 Å resolution) of GlcPse (PDB accession number: 4LDS) by Phyre2 software [Citation37].
The homology model () of DehrP has 12 TMs that are interlinked by six periplasmic loops on the extracellular face and five intracellular loops on the cytoplasmic face of the membrane. However, the large periplasmic loop between TMs 9 and 10 proposed by Tse et al. [Citation21] was not modelled by the Phyre2 server [Citation37], specifically because the template does not have such a loop and the model is comprised of 12 TMs, typical of an MFS transporter. Therefore, removal of the large loop would allow for the modelling of more haloacid transporters transmembrane helices based on the existing experimentally determined 3D structures of MFS transporters in the PDB. The relatively large cytoplasmic α-helical loop connecting TM6 and TM7 () serves as a linker between the two domains (N and C) of the 3D structure of DehrP and is within the 30−100 residues that connect the two domains of MFS transporters [Citation24–30,Citation32]. Like all known 3D structures of MFS transporters, the intracellular loops between TMs 2 and 3, and 8 and 9 of DehrP appear quite short. Their role is presumably to reduce the flexibility of the loops on the cytoplasmic face of the MFS proteins [Citation31]. The two DehrP domains (N and C) are superimposable () with the inward facing structure of GlcPse (template), and the common binding site residues in the cavity of the domains are well aligned.
Figure 2. Overview of DehrP structural arrangement. (A) Side-view representation of the 12 TMs as two-fold pseudosymmetrical domains connected by a relatively large cytoplasmic α-helical loop between TM6 and TM7. (B) Periplasmic-view showing the cavity that leads into the periplasmic space with TM7 at the middle, while TMs 3, 6, 9, and 12 farther away from the cavity. (C) Cytoplasmic-face showing the cavity that leads into the cytoplasm with TM7 at the middle, while TMs 3, 6, 9, and 12 farther away from the cavity.
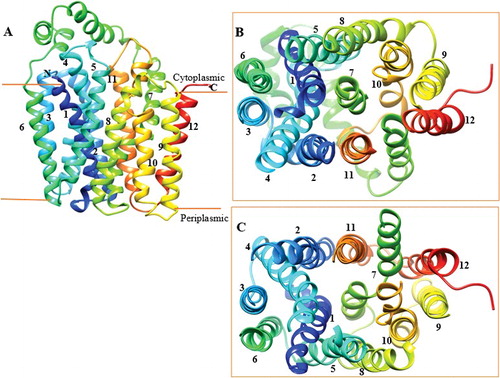
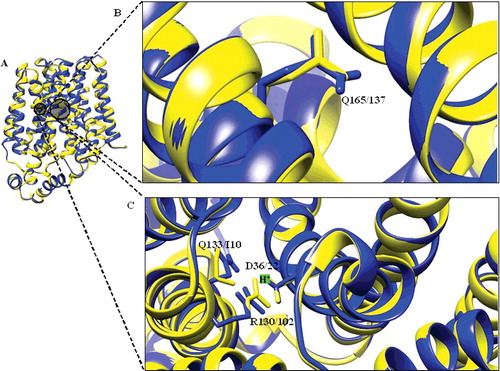
Figure 3. Structural alignment of DehrP. (A) Superposition of DehrP (blue) and GlcPse (yellow) in a schematic representation. (B) Superposition of Gln165 of DehrP on the glucose binding site residue (Gln137) of GlcPse. (C) Gln133, Asp36 and Arg130 of DehrP superposition on the proposed H+-binding site residues (Ile105, Asp22 and Arg102) of GlcPse. The grey box shows the domain region including the catalytic region.
Validity of the 3-D model
The quality and reliability of the DehrP structure was assessed using z-score ProSA-web [Citation39], RAMPAGE [Citation40] Ramachandran plot and ERRAT [Citation41]. The DehrP protein obtained a z-score of −2.86, which is well within the range of values observed for experimentally resolved native protein structures of a similar size (z-score ≤ 10) [Citation39], (supplementary Figure S2). It is worth noting that the calculated protein z-score for DehrP is comparable to scores obtained for PDB proteins whose structures were resolved by X-ray crystallography and nuclear magnetic resonance. The result, therefore, indicates that the obtained model was of good quality. Further, the stereochemical quality (Ф and Ѱ angles) of the model by the Ramachandran plot revealed that most of the residues (93.6%) are in the favoured region (supplementary Figure S3), whereas the remaining 3.9% and 2.4% are in the allowed and outlier regions, respectively. The data indicate that the generated DehrP protein model was good. To check the reliability of the model, we performed a statistical analysis of the non-bonded interactions between different atom types by ERRAT [Citation41]. The ERRAT plot (supplementary Figure S4) shows that the overall quality factor (OQF) of the model is ∼91%. The average OQF of low resolutions (2.5–3 Å) structures is found to be 91% [Citation41], hence affirming the reliability of the obtained 3D protein model.
Putative binding residues and docking analysis
Transporters are membrane proteins that mediate the uptake of substrates into and out of the cells. The transport process is possible because of the presence of key residues that coordinate the passage of substrates across the membrane [Citation50,Citation51]. Based on the sequence similarity, some of these essential residues were found conserved among the characterised MFS transporters [Citation27,Citation52]. Superposition of the 3-D structure of DehrP on that of GlcPse revealed the presence of common binding site residues (). Asp36, Arg130 and Gln133 in DehrP overlapped, respectively, with the H+-binding site residues Ile105, Asp22 and Arg102 in GlcPse, whereas Gln165 overlapped with the glucose-binding site residue Gln137. Studies have shown that these residues play important roles in the transport of H+ into the cells [Citation27,Citation32]. B. caribensis MBA4 haloacid transporters (Deh4p and Dehp2) are reportedly dependent on pH and transmembrane electrochemical gradient for transport activities [Citation1]. Previous comparative analysis showed that DehrP is a member of the MHS as seen in its high sequence similarity with known members of MHS [Citation20]. It is possible that these residues could be the binding sites for H+ during haloacid transport into the bacterial cell, considering that residues Asp36, Arg130 and Gln133 are a part of the DehrP sequence motifs [Citation20]. Conversely, a previous biochemical study revealed that glutamine is important for transport activity and exofacial ligand binding [Citation53]. In this perspective, the Gln165 in DehrP may have an important role in haloacid binding, too.
Docking analyses of the 3D structure of DehrP with substrates, DGlc, MBA, MCA, DBA, DCA, TCA, and 2,2-DCP, show that, in addition to Gln165, there are at least four other possible residues involved in haloacid binding. The complexes with the best conformation were selected from nine docking poses for each ligand. The visual screening of the docking poses showed that residues, Glu33, Trp34, Phe37, Phe38, Gln165 and Glu370 are potential binding sites in the DehrP cavity () and these residues correspond, respectively, to Thr19, Gly20, Asn23, Gly24, Gln137 and Asn256 in GlcPse. It is important to mention here that both Gln137 and Asn256 are among the five known glucose-binding residues in GlcPse () [Citation32]. Other residues, Glu33, Trp34, Phe37 and Phe38 are found on TM1. Gln165 and Glu370 are located on TMs 5 and 7, respectively. Some residues on TMs 1, 4, 5, 7 and 10 have been reported to contribute to substrate binding in the MFS transporters [Citation24–30,Citation32]. It was demonstrated that Glu33, Trp34, Phe37, Phe38 and Gln165 are part of DehrP sequence motifs [Citation20].
Figure 4. Docked conformation of DehrP (model) with DGlc. (A) Putative DehrP binding site in the inward-facing conformation (rainbow) complexed with D-glucose (DGlc), monobromoacetate (MBA), monochloroacetate (MCA), dibromoacetate (DBA), dichloroacetate (DCA), trichloroacetate (TCA) and 2,2-dichloropropionate (2,2-DCP) shown as rectangular grey shading. (B) Expanded view of the binding site residues Glu33, Trp34, Phe37, Phe38, Gln165 and Glu370 (stick model) and DGlc (purple), MBA (yellow), MCA (blue), DBA (green), DCA (pink), TCA (black) and 2, 2-DCP (red) in the ball and stick representation.
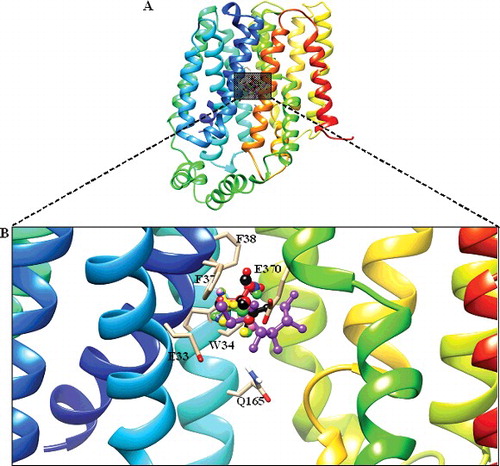
Figure 5. Docked conformation of GlcPse (template). (A) The glucose binding site in the inward-facing conformation (rainbow) complexed with D-glucose (DGlc) and monobromoacetate (MBA), monochloroacetate (MCA) shown as rectangular grey shading. (B) Expanded view of the binding site residues (Gln137, Gln250, Gln251, Asn256 and Trp367) complexed with DGlc (purple), MBA (yellow) and MCA (blue) in the ball and stick representation.
The study observed some disparities in the binding free energy values calculated in AutoDock Vina [Citation46] (see supplementary Table S1) for seven DehrP–ligands complexes. The best docked conformation and affinity of the DehrP–DGlc complex is comparable to that of the GlcPse–DGlc complex. The DehrP–MBA and DehrP–MCA complexes showed the lowest binding affinity which correlates well with the fact that the two ligands are less substituted with halogen and both are not substrates for growth of Rhizobium sp. RC1. The GlcPse–MBA and GlcPse–MCA complexes showed similar affinity, whereby an increase in the number of halogens leads to an increase in the affinity of DehrP for the ligands. The study believes the halogens formed weak interactions with the binding site residues as illustrated in . Halogen bonds (C−X···D−Z) between halogenated ligands and binding-site residues containing polar nitrogen and oxygen atoms can significantly alter the ligand-binding affinity to the target protein receptor [Citation54], depending on the lipophilicity substituent constant (π) of the halogen [Citation55]. The higher affinity of DehrP for DBA as compared to DCA might be due to the higher lipophilicity substituent constant (π) of the bromine atom. Docking of DCA, DBA, TCA and 2,2-DCP with GlcPse showed that the ligand could not interact with the glucose binding site residues. This is consistent with the fact that GlcPse is not a transporter of haloacids, attributable to differences in the binding site residues in DehrP and GlcPse. The presence of electron-rich halogens in haloacids and the halogen bond-acceptor side chains in DehrP (Trp34 Phe37, Phe38, Glu33 and Glu370) may have contributed to the greater affinity of DehrP for haloacids. This is consistent with reports by previous works which described the affinity of halogens for O and aromatic rings [Citation56–58]. Among all the haloacids docked with DehrP, 2,2-DCP showed the highest binding affinity, consistent with the fact that 2,2-DCP is the natural substrate for growth of Rhizobium sp. RC1. The DehrP binding site conformations were further analysed using Discovery Studio v.16.1.0 [Citation47] and it was revealed that the haloacid ligands predominantly interact with the DehrP binding site residues through van der Waals interactions (). In addition to the six putative binding site residues (Glu33, Trp34, Phe37, Phe38, Gln165 and Glu370), it appears that the Ala165 result was somewhat anticipated, as involvement of Leu and Ala in halogen bond formation is well reported in the literature [Citation59]. As a matter of fact, Leu is considered to be an important halogen interaction initiator [Citation60].
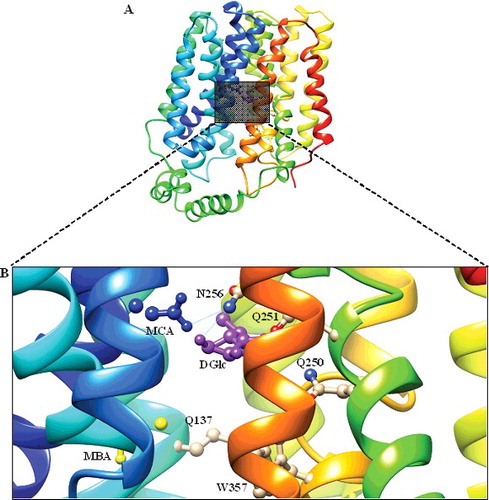
Figure 6. Illustration of halogen bonds and aromatic interactions in DehrP-haloacid complexes. The ligands and binding site residues are shown in the ball and stick presentation. The protein is presented as a cartoon model and coloured by atom type with carbon atom in grey, hydrogen in white, oxygen in red, nitrogen in blue, chloride in green and bromide in purple. (A) Monobromoacetate (MBA in yellow) ligand forming Br···aromatic ring interaction with Trp34. (B) Monochloroacetate (MCA in blue) ligand forming Cl···aromatic ring interaction with Phe38. (C) Dibromoacetate (DBA in green) ligand forming Br···aromatic ring interactions with Trp34 and Phe38. (D) Dichloroacetate (DCA in pink) ligand forming Cl···aromatic ring interactions with Trp34 and Phe38. (E) Trichloroacetate (TCA in black) ligand forming Cl···aromatic ring interactions with Trp34, Phe37 and Phe38. (F) 2,2-dichloropropionate (2,2-DCP in red) ligand forming Cl···aromatic ring interactions with Trp34 and Phe38.
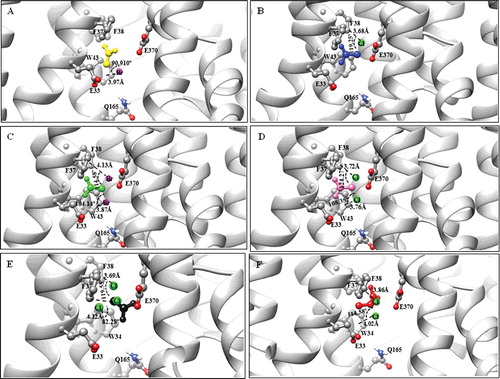
Figure 7. Overall DehrP interacting residues with haloacid ligands.
Proposed haloacid transport mechanism
Based on the in silico data, we herein propose the mechanism of haloacid transport for DehrP via the previously described alternative access transport model of E. coli lactose permease (LacY) [Citation24] and GlcPse glucose permease [Citation32]. In this mechanism, the sugar- and H+-binding sites are alternately exposed to either side of the membrane ((A) I-III). Unlike LacY and GlcPse, the side chains that are important for haloacid- and H+- binding in DehrP are usually located in the N-terminal helix bundle ((B)). In the absence of H+, Asp36 and Arg130 form a salt bridge that consequently leads to the widening of the binding cavity following the shift in positions of TMs 1 and 4 ((B) IV). When H+ is present, Asp36 is protonated and the salt bridge is disrupted. This causes the TMs 1 and 4 to undergo rearrangement which decreases the size of the binding cavity ((B) V). Most importantly, the smaller binding cavity results in a concomitant lowering of the energetic barrier of the transporter's conformations [Citation32], hence permitting the rapid translocation of the haloacid ((B) VI). The N- and C-domains are also brought closer when the haloacids and residues between the two domains interact ((B) VI).
Figure 8. Proposed mechanism of haloacid:H+ Symport. (A) Transport cycle of DehrP shows the postulated steps with the outward-facing (I), and inward-facing (II) conformations separated by the occluded intermediate conformation (III). Haloacid (S; 2,2-DCP) and proton (H+; +) are coupled in the process. (B) Salt bridge forms between the side chains of Asp36 (-COO−) and Arg130 (-NH3+) in the absence of H+ (IV). Protonated D36 shifts closer to the Asp36 binding cavity (V). H+ and S are coordinated by the binding residues in the cavity (VI). The genomic localization of dehrP with dehalogenase genes (dehD, dehL and dehE) and their upstream promoters (P1 and P2) was drawn from [Citation15,Citation61].
![Figure 8. Proposed mechanism of haloacid:H+ Symport. (A) Transport cycle of DehrP shows the postulated steps with the outward-facing (I), and inward-facing (II) conformations separated by the occluded intermediate conformation (III). Haloacid (S; 2,2-DCP) and proton (H+; +) are coupled in the process. (B) Salt bridge forms between the side chains of Asp36 (-COO−) and Arg130 (-NH3+) in the absence of H+ (IV). Protonated D36 shifts closer to the Asp36 binding cavity (V). H+ and S are coordinated by the binding residues in the cavity (VI). The genomic localization of dehrP with dehalogenase genes (dehD, dehL and dehE) and their upstream promoters (P1 and P2) was drawn from [Citation15,Citation61].](/cms/asset/f9afb628-cd71-45ba-b0ef-235a7364d633/tbeq_a_1432417_f0008_oc.jpg)
Conclusions
This work demonstrates that haloacid transport proteins are implicated in the coordination of haloacids from the environment into the cells of Rhizobium sp. RC1 during haloacid degradation. The results from this study represent a target for improved haloacid uptake. The 3-D structure shows that DehrP is MFS that can be folded into 12 TMs, constructed by two-fold pseudosymmetrical domains connected by a relatively large cytoplasmic loop between TMs 6 and 7. Docking analysis with haloacid ligands revealed that the halide atoms and carboxylic groups in the ligands typically participated in the formation of halogen bonds and van der Waals interactions through residues Glu33, Trp34, Phe37, Phe38, Gln165 and Glu370 of the DehrP protein. The study also revealed that residues Ala165 and Leu373 could be important in protein–ligand interactions and that the transport of haloacid possibly occurs via coupling of the compound with H+ transport through the H+-binding site residues Asp36, Arg130 and Gln133. Based on the well-studied LacY and GlcPse models, we propose that the DehrP haloacid transport in Rhizobium sp. RC1 takes place via the alternative access mechanism of substrate transport by the MFS proteins. Hence, the present work was successful in identifying the haloacid binding site on DehrP. The data obtained in this study can be used for future work involving site-directed mutagenesis on haloacid transporters to enhance transport of haloacids from the environment into bacterial cells for improved rate of haloacid degradation.
Supplemental_Data.pdf
Download PDF (653.1 KB)Acknowledgments
M.A. Musa thanks the Nigerian Goverment for the Studenship award 2014/2015.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Su X, Kong K-F, Tsang JSH. Transports of acetate and haloacetate in Burkholderia species MBA4 are operated by distinct systems. BMC Microbiol. 2012; [cited 2017 Oct 16];12(1):267. [8 p.] DOI:10.1186/1471-2180-12-267
- Su X, Deng L, Kong KF, et al. Enhanced degradation of haloacid by heterologous expression in related Burkholderia species. Biotechnol Bioeng. 2013;110(10):2687–2696.
- Tsang JSH, Sallis PJ, Bull AT, et al. A monobromoacetate dehalogenase from Pseudomonas cepacia MBA4. Arch Microbiol. 1988;150(5):441–446.
- Stringfellow JM, Cairns SS, Cornish A, et al. Haloalkanoate dehalogenase II (DehE) of a Rhizobium sp. — molecular analysis of the gene and formation of carbon monoxide from trihaloacetate by the enzyme. Eur J Biochem. 1997;250(3):789–793.
- Effendi AJ, Greenaway SD, Dancer BN. Isolation and characterization of 2,3-dichloro-1-propanol-degrading rhizobia. Appl Environ Microbiol. 2000;66(7):2882–2887.
- Berthiaume C, Gilbert Y, Fournier-Larente J, et al. Identification of dichloroacetic acid degrading Cupriavidus bacteria in a drinking water distribution network model. J Appl Microbiol. 2014;116(1):208–221.
- van der Ploeg J, Janssen DB. Sequence analysis of the upstream region of dhlB, the gene encoding haloalkanoic acid dehalogenase of Xanthobacter autotrophicus GJ10. Biodegradation. 1995;6(3):257–263.
- Berry EKM, Allison N, Skinner AJ, et al. Degradation of the selective herbicide 2,2-dichloropropionate (Dalapon) by a soil bacterium. Microbiol. 1979;110(1):39–45.
- Janssen DB, Scheper A, Dijkhuizen L, et al. Degradation of halogenated aliphatic compounds by Xanthobacter autotrophicus GJ10. Appl Environ Microbiol. 1985;49(3):673–677.
- Chaudhry GR, Chapalamadugu S. Biodegradation of halogenated organic compounds. Microbiol Rev. 1991;55(1):59–79.
- Strotmann UJ, Pentenga M, Janssen DB. Degradation of 2-chloroethanol by wild type and mutants of Pseudomonas putida US2. Arch Microbiol. 1990;154(3):294–300.
- Plewa MJ, Simmons JE, Richardson SD, et al. Mammalian cell cytotoxicity and genotoxicity of the haloacetic acids, a major class of drinking water disinfection by-products. Environ Mol Mutagen. 2010;51(8–9):871–878.
- Higgins TP, Hope SJ, Effendi AJ, et al. Biochemical and molecular characterisation of the 2,3-dichloro-1-propanol dehalogenase and stereospecific haloalkanoic dehalogenases from a versatile Agrobacterium sp. Biodegradation. 2005;16(5):485–492.
- Yu M, Faan YW, Chung WY, et al. Isolation and characterization of a novel haloacid permease from Burkholderia cepacia MBA4. Appl Environ Microbiol. 2007;73(15):4874–4880.
- Jing N, Wahab RA, Hamdan S, et al. Cloning and DNA sequence analysis of the haloalkanoic permease uptake gene from Rhizobium sp. RC1. Biotechnology. 2010;9(3):319–325.
- Su X, Tsang JS. Existence of a robust haloacid transport system in a Burkholderia species bacterium. Biochim Biophys Acta. 2013;1828(2):187–192.
- Leigh J, Skinner A, Cooper RA. Isolation and partial characterisation of dehalogenase-deficient mutants of a Rhizobium sp. FEMS Microbiol Lett. 1986;36(2–3):163–166.
- Leigh JA, Skinner AJ, Cooper RA. Partial purification, stereospecificity and stoichiometry of three dehalogenases from a Rhizobium species. FEMS Microbiol Lett. 1988;49(3):353–356.
- Sudi IY, Shamsir MS, Jamaluddin H, et al. Interactions of non-natural halogenated substrates with D-specific dehalogenase (DehD) mutants using in silico studies. Biotechnol Biotechnol Equip. 2014;28(5):949–957.
- Musa MA. Computational analysis of putative haloalkanoic permease (DehrP) from Rhizobium sp. RC1. [Master's thesis]. Johor Bahru: Universiti Teknologi Malaysia; 2017.
- Tse YM, Yu M, Tsang JS. Topological analysis of a haloacid permease of a Burkholderia sp. bacterium with a PhoA-LacZ reporter. BMC Microbiol. 2009; [cited 2017 Oct 16];9(1):233. [12 p.] DOI: doi:10.1186/1471-2180-9-233
- Saier MH, Reddy VS, Tsu BV, et al. The transporter classification database (TCDB): recent advances. Nucleic Acids Res. 2016;44(database issue):D372–D379.
- Abramson J, Kaback HR, Iwata S. Structural comparison of lactose permease and the glycerol-3-phosphate antiporter: members of the major facilitator superfamily. Curr Opin Struct Biol. 2004;14(4):413–419.
- Abramson J, Smirnova I, Kasho V, et al. Structure and mechanism of the lactose permease of Escherichia coli. Science. 2003;301(5633):610–615.
- Huang Y, Lemieux MJ, Song J, et al. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science. 2003;301(5633):616–620.
- Yin Y, He X, Szewczyk P, et al. Structure of the multidrug transporter EmrD from Escherichia coli. Science. 2006;312(5774):741–744.
- Dang S, Sun L, Huang Y, et al. Structure of a fucose transporter in an outward-open conformation. Nature. 2010;467(7316):734–738.
- Newstead S, Drew D, Cameron AD, et al. Crystal structure of a prokaryotic homologue of the mammalian oligopeptide-proton symporters, PepT1 and PepT2. EMBO J. 2011;30(2):417–426.
- Solcan N, Kwok J, Fowler PW, et al. Alternating access mechanism in the POT family of oligopeptide transporters. EMBO J. 2012;31(16):3411–3421.
- Sun L, Zeng X, Yan C, et al. Crystal structure of a bacterial homologue of glucose transporters GLUT1-4. Nature. 2012;490(7420):361–366.
- Yan N. Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem Sci. 2013;38(3):151–159.
- Iancu CV, Zamoon J, Woo SB, et al. Crystal structure of a glucose/H+ symporter and its mechanism of action. Proc Natl Acad Sci USA. 2013;110(44):17862–17867.
- The Uniprot Consortium : the universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158–D169.
- Altschup SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410.
- Schaffer AA, Aravind L, Madden TL, et al. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 2001;29(14):2994–3005.
- Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011 [cited 2017 Oct 16];7(1):539. [6 p.] DOI:10.1038/msb.2011.75
- Kelley LA, Mezulis S, Yates CM, et al. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–858.
- Bhattacharya D, Nowotny J, Cao R, et al. 3Drefine: an interactive web server for efficient protein structure refinement. Nucleic Acids Res. 2016;44(W1):W406–409.
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35(web server issue):W407–W410.
- Lovell SC, Davis IW, Arendall WB, et al. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;50(3):437–450.
- Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Prot Sci. 1993;2(9):1511–1519.
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612.
- Meredith D, Price R. Molecular modeling of PepT1—towards a structure. J Membr Biol. 2006;213(2):79–88.
- Pence HE, Williams A. ChemSpider: An online chemical information resource. J Chem Edu. 2010;87(11):1123–1124.
- Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791.
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31(2):455–461.
- Dassault Systèmes BIOVIA. Discovery studio modeling environment, release 2017. San Diego (CA): Dassault Systèmes; 2016.
- Liò P, Bishop M. Modeling sequence evolution. Bioinformatics. 2008; 452: 255–285.
- Shenoy SR, Jayaram B. Proteins: sequence to structure and function-current status. Curr Prot Pep Sci. 2010;11(7):498–514.
- Hashiramoto M, Kadowaki T, Clark AE, et al. Site-directed mutagenesis of GLUT1 in helix 7 residues 282 results in perturbation of exofacial ligand binding. J Biol Chem. 1992;267(25):17502–17507.
- Mueckler M, Makepeace C. Model of the exofacial substrate-binding site and helical folding of the human Glut1 glucose transporter based on scanning mutagenesis. Biochemistry. 2009;48(25):5934–5942.
- Sanderson NM, Qi D, Steel A, et al. Effect of the D32N and N300F mutations on the activity of the bacterial sugar transport protein, GalP. Biochem Soc Trans. 1998;26(3):S306.
- Mueckler M, Weng W, Kruse M. Glutamine 161 of Glut1 glucose transporter is critical for transport activity and exofacial ligand binding. J Biol Chem. 1994;269(32):20533–20538.
- Olsen J, Seiler P, Wagner B, et al. A fluorine scan of the phenylamidinium needle of tricyclic thrombin inhibitors: effects of fluorine substitution on pKa and binding affinity and evidence for intermolecular C-F…CN interactions. Org Biomol Chem. 2004;2(9):1339–1352.
- Bois F, Beney C, Boumendjel A, et al. Halogenated chalcones with high-affinity binding to P-glycoprotein: Potential modulators of multidrug resistance. J Med Chem. 1998;41(21):4161–4164.
- Voth AR, Ho PS. The role of halogen bonding in inhibitor recognition and binding by protein kinases. Curr Top Med Chem. 2007;7(14):1336–1348.
- Matter H, Nazare M, Gussregen S, et al. Evidence for C-Cl/C-Br…pi interactions as an important contribution to protein-ligand binding affinity. Angew Chem Int Ed Engl. 2009;48(16):2911–2916.
- Hardegger LA, Kuhn B, Spinnler B, et al. Systematic investigation of halogen bonding in protein–ligand interactions. Angew Chem Int Ed. 2011;50(1):314–318.
- Sirimulla S, Bailey JB, Vegesna R, et al. Halogen interactions in protein–ligand complexes: implications of halogen bonding for rational drug design. J Chem Inf Mod. 2013;53(11):2781–2791.
- Wilcken R, Liu X, Zimmermann MO, et al. Halogen-enriched fragment libraries as leads for drug rescue of mutant p53. J Am Chem Soc. 2012;134(15):6810–6818.
- Adamu A, Wahab RA, Huyop F. L-2-Haloacid dehalogenase (DehL) from Rhizobium sp. RC1. SpringerPlus. 2016; [ cited 2017 Oct 16];5(1):695. [17 p.] DOI:10.1186/s40064-016-2328-9