
Abstract
To identify the amino acid residues that are the critical factors contributing to the high catalytic activity and good thermostability of Yersinia enterocolitica subsp. palearctica non-specific nuclease (Y. NSN), in this work, we analyzed the structure and the functional domain of Y. NSN by using bioinformatics software. Disulphide bonds were analyzed by mass spectrometry. Six active site mutants of Y. NSN, including two salt bridge mutants and four disulphide bond mutants, were obtained by site-directed mutagenesis. Their enzyme activity and thermostability were assayed. The results showed that when changing the amino acids of the active site, or breaking the salt bridges and disulphide bonds, the enzyme activity and thermal stability of Y. NSN decreased significantly, compared to those of the wild-type enzyme. It is indicated that the active site, salt bridges and disulphide bonds play an important role in the enzyme activity and thermostability of Y. NSN.
Introduction
Non-specific nucleases are a group of enzymes that degrade both single- and double-stranded DNA and RNA without sequence specificity [Citation1–3]. They are detected from a wide variety of sources, such as virus, bacteria, fungi and animals [Citation4–7]. They are involved in nucleic acid metabolism, as well as in a range of cell processes, such as DNA repair, DNA restriction, replication, recombination, transposition, transcription and topoisomerization and RNA processing, splicing, editing and interference [Citation1]. In addition, they have been widely used in avoiding mutation, nucleoside deletion, phosphoric acid removal in growth and metabolism processes, host defenses against the invasion of exogenous nucleic acid molecules, and cell apoptosis [Citation8–16]. Presently, the studies on non-specific nucleases have been focused mostly on Serratia marcescens nuclease: its gene regulation, protein secretion, endonuclease action and protein structure. The production, expression, extracellular secretion, catalytic mechanism, structure and application of S. marcescens nucleases were studied by Benedik and Strych [Citation17] and Mahua et al. [Citation18]. However, a few studies on other nucleases, such as Staphylococcus nucleases, onconase, sugar non-specific nucleases and nuclease P1 have been reported [Citation19–23]. A non-specific nuclease from Yersinia enterocolitica subsp. palearctica (Y. NSN) was found by our group. According to the sequence alignment, Y. NSN showed high similarity to S. marcescens nucleases. Y. NSN had similar enzyme activity compared to S. marcescens nucleases at the same conditions [Citation9, Citation10]. Therefore, we speculated that they may also show similar features in their structure and function.
In our previous work, a recombinant plasmid containing the Y. NSN gene and PET-24a(+) was expressed in BL21starTM(DE3)plysS. Our results showed that Y. NSN had high enzyme activity, good thermostability and wide resistance to acid conditions, alkaline conditions, temperature and various chemical substances [Citation9]. Therefore, on the basis of structure analysis and prediction of active sites [Citation10, Citation24], in order to identify the amino acid residues that are the critical factors that contribute to the high catalytic activity of Y. NSN, Y. NSN mutants were obtained by site-directed mutagenesis in this study. It has been reported that salt bridges play an important role in the high temperature tolerance of the protein. There are more salt bridges in thermophilic proteins, especially in extremely thermophilic proteins [Citation25, Citation26]. Serratia marcescens nuclease has two pairs of disulphide bonds [Citation17]. A mutant in one of these disulphide bonds showed only one in tenth of the wild-type (WT) enzyme activity and a ten thousandth of its thermostability, which indicated that the disulphide bond played an important role in the thermostability of the nuclease [Citation27–29]. We also observed a significant decrease in the enzyme activity and thermostability of Y. NSN after breaking its salt bridge and disulphide bond. The enzyme activity and thermostability of the mutants and WT enzyme were compared. Moreover, the structure of Y. NSN was analyzed to interpret the effect of the mutations on the enzyme activity and thermostability of Y. NSN. The results from this work contribute to uncovering the factors involved in enzyme activity and thermostability of Y. NSN.
Materials and methods
Materials
StarPrep Plasmid Miniprep Kit, 2 × Pfu PCR StarMix Loading Dye-free, 2 × Pfu PCR StarMix and StarPrep Gel Extraction Kit were purchased from GenStar (Beijing, China). Calf thymus DNA was obtained from Sigma (Shanghai, China). Isopropyl-β-D-thiogalactopyranoside (IPTG), restriction endonucleases NdeI and XhoI, T4 DNA Ligase, kanamycin sulphate, Methylase Dpn I, BCA Protein Quantization Kit and all other chemicals (analytical grade purity) were purchased from Sangon (Shanghai, China). The primer synthesis and mutant DNA sequencing were conducted by Sunny (Shanghai, China).
Structural analysis
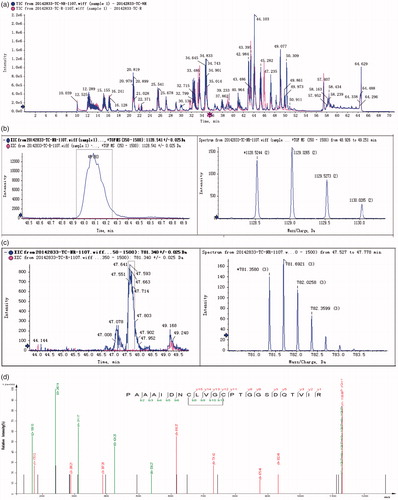
The functional domains of Y. NSN were analyzed by CDD (https://www.ncbi.nlm.nih.gov/cdd/). The salt bridges of Y. NSN were analyzed by Accelry Discovery Studio.v.2.5. The amino acid sequence alignment between Y. NSN and S. marcescens nucleases was analyzed by Clustal X. The results from the sequence alignment were presented in colour by BoxShade (https://embnet.vital-it.ch/software/BOX_form.html).
Mass spectrometry analysis
Mass spectrometry was used to verify the existence of the disulphide bond in Y. NSN. First, the sample was subjected to enzymolysis and reduction reaction. Then, 2.0 nmol sample and ddH2O were mixed in order to obtain the 100 μL mixture, 100 μL urea (UA), 10 μL iodoacetamide (IAM) (1.0 mol/L) and 3.0 μg lysine were added into the mixture and mixed well, and then the reaction system was stored in 37 °C for 3 h. Next, 300 μL NH4HCO3 (25 mmol/L), 6.0 μg tryptophan/chymotrypsin were added into the mixture and the reaction system was stored at 37 °C for 20 h. Dithiothreitol (DTT, 20 mmol/L) was added to the reaction system for the reducing reaction. After the reaction was started at 56 °C for 30 min, 100 mmol/L IAM was then mixed away from light and preserved for 20 min.
HPLC (high performance liquid chromatography) was used to separate the enzymolysis and reduction products. An LC-20AD liquid phase system was used. The mobile phase was held at 100% buffer A (0.10% formic acid), followed by a linear gradient from 4.0 to 70% buffer B (0.10% formic acid in 90% acetonitrile) over 70 min with a flow rate of 400 μL/min. An Agilent Zorbax SB C18 column was used with the balance of buffer A. The samples were injected into the column by an auto-feeding device and then separated by the chromatography column. After separation and desalination by capillary HPLC, the sample was then tested and analyzed by a TripleTOF™ 5600+ mass spectrometer (AB SCIEX) for 70 min. The MS scan(m/z 350–3000)was followed at 150 cycles per second with 40,000 resolution. The first mass spectrometry data of peptide fragments which have located disulphide bonds were found in the data base; the secondary spectrogram of the peptide fragments with located disulphide bonds were verified in order to confirm the linking mode of the disulphide bonds.
Mutation design
In order to identify the amino acid residues that are the critical factors for the high catalytic activity of Y. NSN, the six predicted active sites were mutated into Ala. Six active site mutants, D124A, R125A, H127A, N157A, E165A and R169A, were obtained by site-directed mutagenesis. Likewise, to identify the critical factors that contribute to the high thermostability of Y. NSN, Arg63 in the salt bridge Arg63–Asp100 and Asp166 in the salt bridge Asp166–Arg169 were mutated into Gln, respectively. We eliminated some of the salt bridges in Y. NSN, so Y. NSN salt bridge subtraction mutants R63Q and D166Q were obtained. In addition, each Cys that participates in forming a disulphide bond in Y. NSN was mutated into Ser. Four disulphide bond mutants, C48S, C52S, C238S and C252S, were obtained by site-directed mutagenesis.
Site-directed mutagenesis
A recombinant plasmid PET-24a (+) containing the Y. NSN gene was used to clone Y. NSN. Site-directed mutagenesis of the Y. NSN gene used in this study was performed using the quick-change site-directed method described by Fisher and Pei [Citation30]. All primers are listed in . A final concentration of 0.50 μmol/L of each pair of complementary primers and 5.0 ng of plasmid DNA as a template were used for amplification. The 50 μL PCR reaction mixture included 25 μL of 2 × Pfu PCR StarMix and 22 μL of ddH2O. The directed mutagenesis PCR program was performed under the following conditions: 95 °C for 30 s; 15 cycles of 95 °C for 30 s, 1 min at X °C for annealing, 6 min at 72 °C for extension (X represents the annealing temperature and was determined by the Tm of the primers); with a final extension at 72 °C for 5 min. To eliminate the effect of the PCR ingredients on the band and to obtain the desired products, the PCR products were purified by a StarPrep Gel Extraction Kit. A 1.0-mL sample of PCR product was digested with 10 U of Dpn I at 37 °C for 1.0 h. The reaction mixtures were transformed into DH5α Escherichia coli competent cells. The successful introductions of the desired mutations were verified by sequencing at Sunny (Shanghai, China). Twelve mutants that were confirmed by DNA sequencing were obtained for this study.
Table 1. Primers used for mutations.
Construction of expression vector
The 852 bp coding region of Y. NSN was amplified by the PCR method. The PCR reaction mixture (20 μL) included 1.0 μL of plasmid DNA, 1.0 μL of each primer (10 μmol/L), 10 μL of 2 × Taq PCR StarMix and 7.0 μL of ddH2O. The PCR program was performed as follows: 94 °C for 2 min; 30 cycles of 94 °C for 30 s, 30 s at 58 °C for annealing, 5 min at 72 °C for extension; 72 °C for 5 min; and finally held at 4 °C. The PCR reaction products were then subjected to 1.0% agarose gel electrophoresis. According to the instructions of the DNA gel extraction kit, the PCR reaction products were purified and recovered. The PCR reaction products and the recombinant plasmid containing the Y. NSN gene and PET-24a (+) were subjected to the double digests by the restriction endonucleases NdeI and XhoI, respectively. The 50 μL reaction mix of the PCR reaction products’ double digestion contained 20 μL of the PCR reaction products, 5.0 μL of NEB buffer, 2.5 μL of the Nde I (10 U/μL), 2.5 μL of the Xho I (10 U/μL), 5.0 μL of BSA (1.0 mg/mL) and 15 μL of ddH2O. The 50 μL reaction mix of the vector’s double digestion contained 30 μL of the PET-24a vector, 5.0 μL of NEB buffer, 2.5 μL of the Nde I (10 U/μL), 2.5 μL of the Xho I (10U/μL), 5.0 μL of BSA (1.0 mg/mL) and 5.0 μL of ddH2O. The 50 μL reaction mix was stored at 37 °C for 3–4 h and then subjected to 1.0% agarose gel electrophoresis. According to the instructions of the StarPrep Gel Extraction Kit, the double digest reaction products were purified and recovered. By using the T4 DNA Ligase and storing at 16 °C for 2–3 h, the recombinant plasmid containing the Y. NSN gene and PET-24a was constructed as the expression vector. The 20 μL reaction mix contained 2.0 μL of 10x T4 DNA Ligase buffer, 3.0 μL of the plasmid and 14 μL of the PCR products after the enzyme digestion and 1.0 μL of T4 DNA Ligase. Finally, this engineered vector was transformed into the expression host. Three transformants of each mutant were selected randomly and sequenced to ensure correct substitution mutations.
Enzyme expression
The recombinant plasmids PET-24a (+) containing the Y. NSN genes were transformed into BL21starTM(DE3)plysS competent cells. For each mutant, 15 colonies were selected. The recombinant cells were grown in Luria–Bertani (LB) medium containing 50 μg/mL of kanamycin sulphate at 37 °C. Ten milliliters of overnight pre-culture was seeded into 20 mL of fresh LB medium containing 50 μg/mL of kanamycin sulphate and incubated at 37 °C and 200 rpm to an optical density at 600 nm (OD600) of 0.8–1.0. Following induction with a final concentration of 1.0 mmol/L IPTG for another 22 h with shaking at 200 rpm, the cells were harvested by centrifugation at 8000×g for 5 min at 4 °C and re-suspended in 2.0 mL of distilled water supplemented with 20 mmol/L MgCl2. The supernatant of the cell lysate (3–5 cycles of freezing at 20 °C and thawing at 50 °C) was used for the study.
Protein content assay
The protein content of the mutants was determined by a BCA Protein Quantitation Kit. According to the the manufacturer’s instructions, a BCA working solution and BCA protein standard solution (0.50 mg/mL) were prepared and 1.0 mL BCA working solution was added to each tube with protein standard solution separately. Each mixture was stored in 60 °C for 30 min after sufficient mixing. Afterwards, the absorption (optical density, OD) at 562 nm was measured after the samples were restored at room temperature. The standard equation is Y = 1822.6X–7.2015, R2=0.9929. Then, 10 μL of sample and 90 μL PBS were added into a 1.5-mL centrifuge tube and the absorption at 562 nm wavelength was measured according to the method described previously. The protein content of the sample was calculated using the protein standard curve. The solution was concentrated with the BCA protein assay kit to approximately 5.0 mg/mL prior to the following experiments ().
Table 2. Protein content in the supernatant of the cell lysate.
Enzyme activity assay
To analyze the enzyme activity of the mutants, the activity of the WT enzyme was used as a control. The enzyme activity of the mutants and the control were determined according to the method described previously by our group [Citation9]. Briefly, approximately 2.0 μL of diluted enzyme solution was incubated in 38 μL of calf thymus DNA (100 ng/μL) as a substrate. After incubation at 55 °C for 3 min, the reaction was stopped by adding 8.0 μL of 6× DNA loading buffer (10 mmol/L Tris-HCl, 60 mmol/L EDTA, 40% sucrose, 0.050% bromophenol blue, pH7.6). The reaction products were then subjected to 1.0% agarose gel electrophoresis. The degree of nucleic acid degradation was visualized and calculated by staining with 0.25 μg/mL ethidium bromide. In the thermostability assay, the enzymes were incubated at 80 °0 for 30 min, and the residue enzyme activity was measured as above.
Results and discussion
Active sites analysis
Using bioinformatics tools, we analyzed the primary structure, the secondary structure, the domain structure and the tertiary structure of Y. NSN. This work provided the theoretical basis for the site-directed mutagennesis study of Y. NSN. The functional domains of Y. NSN were analyzed by CDD. As shown in , Y. NSN has two functional domains, NUC superfamily (48…274) and Endonuclease_NS (63…263). The active sites of Y. NSN are pos. 124, 125, 127, 157, 165 and pos. 169 [Citation10]. The Mg2+ binding site is pos.157, and the substrate binding sites are pos. 124, 125 and pos. 129. Gao [Citation31] studied the structure and catalytic mechanism of DEDD nuclease from Agrobacterium tumefaciens, in which the divalent metal ions, Mg2+ and Mn2+ were a common cofactor for nucleases. To identify the amino acid residues that are the critical factors that contribute to the high enzyme activity of Y. NSN, mutants of Y. NSN were obtained by site-directed mutagenesis, and their enzyme activity was assayed in the present study.
Figure 1. Prediction of functional domains of Y. NSN by CDD.

The active site of the nuclease is characterized by several conserved residues. In the Serratia nuclease, the conserved residues are Arg-57, Arg-87, His-89, Asn-119, Glu-127 and Arg-131. The role of these residues, in particular His-89 and Asn-119, in the cleavage mechanism, has been analyzed by a detailed mutational and structure analysis. Previously, Frisdhoff [Citation28] reported the common catalytic mechanism of several nucleases. A histidine residue acted as a general base to activate the attacking water and a Mg2+ ion bound to an asparagine residue served as Lewis acid to stabilize the transition state and the leaving group [Citation32].
Enzyme activity analysis
We targeted the six catalytic residues of Y. NSN, Asp124, Arg125, His127, Asn157, Glu165 and Arg169, by mutational analysis to identify the amino acid residues directly involved in catalysis and propose a mechanism of action for this enzyme [Citation10]. To determine the enzyme activity of the WT enzyme and the mutants, we used calf thymus DNA as a substrate. The enzyme activity of the mutants was compared to that of the WT enzyme, which was used as a control (). After incubation, the final reaction products were subjected to 1.0% agarose gel electrophoresis. As the control, distilled water was used instead of the same volume of enzyme. There was almost no degradation of the substrate, which indicated that the control had no enzyme activity. To the contrary, the WT enzyme degraded the substrate into small fragments that were not visible on the gel, which indicated that the WT enzyme retained all or almost all of the enzyme activity. The shorter the fragment in the gel is, the more residual enzyme activity the mutant has.
Figure 2. Effect of active site on enzyme activity of Y. NSN. (a) Control: no nuclease; Lane 0: WT; Lane 1: D124A; Lane 2: R125A; Lane 3: H127A; Lane 4: N157A; Lane 5: E165A; Lane 6: R169A. (b) Enzyme activity of active site mutants.
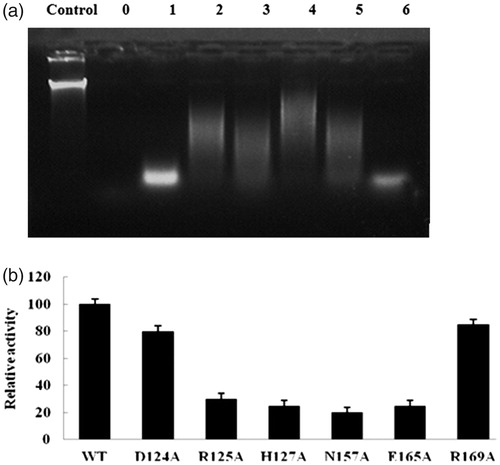
shows the residual enzyme activities of the mutants as a percentage of the activity of the WT enzyme. The R169A and D124A mutants maintained about 85% and 80% of the WT enzyme activity, whereas the R125A, H127A, 157A and E165A mutants only retained about 30, 25, 20 and 25% of it, respectively. Mutations near the catalytic residues most likely influenced the catalytic mechanism [Citation33–35]. Based on the results obtained, the Y. NSN active site mutants D124A, R125A, H127A, N157A, E165A and R169A showed lower enzyme activity than the WT enzyme, especially N157A, which showed the lowest enzyme activity among these six mutants.
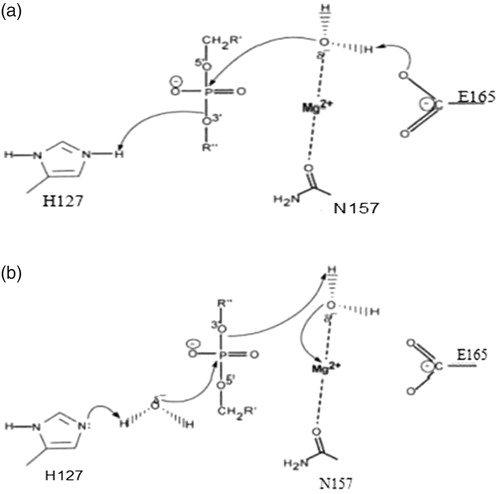
Among these active site residues, His127 and Glu165 appeared to be the general base, whereas Asp124, Arg125 and Arg169 were substrate-binding sites that formed a minor groove that was responsible for binding DNA or RNA [Citation10]. In addition, based on the common catalytic mechanism and the enzyme activity of active site mutants of Y. NSN, the following mechanisms for the hydrolysis of DNA or RNA by Y. NSN are proposed: in the acid catalytic model, the His127 residue acts as a general base to protonate other groups and Glu acts as the general base (); in the base catalytic model, His127 acts as the general base to gain a proton reserved in H2O, H2O is activated in order to attack the ester bond around the phosphorous atom. Besides, Asn157 and a Mg2+ ion play the role of stabilizing the reaction transition state (). Therefore, we suggest that Asn157 was the binding site of Mg2+. As the supplementary factor, Mg2+ plays the most important role in the catalytic mechanism of Y. NSN.
Figure 3. General catalytic model of Y. NSN. (a) General acid model of Y. NSN.His127 residue acting as a general base and protonating other groups and Glu acting as the general base. (b) General base model of Y. NSN.His127 acting as the general base and gaining a proton that H2O reserved; H2O is activated in order to attack the ester bond around the phosphorous atom; Asn157 and Mg2+ ion stabilize the reaction transition state.
Salt bridges analysis
There are six salt bridges in Y. NSN, Asp166-Arg169, Lys209-Glu168, Glu189-Lys250, Lys270-Glu248, Arg63-Asp100 and Asp228-Arg190 [Citation10]. Different salt bridges have different amounts of salt bonds, and in addition, the length of these bonds is also different. As shown in our previous paper [Citation10], the salt bridges Arg63-Asp100 and Asp228-Arg190 have four salt bonds. There is only one salt bond in the salt bridges Asp166-Arg169 and Lys209-Glu168. Through site-directed mutagenesis, the salt bridges were expected to have increased in theory and Y. NSN salt bridge subtraction mutants R63Q and D166Q were obtained successfully.
Fei et al. [Citation26] study elucidated the importance of salt bridges for E. coli AppA phytase’s thermostability and proved that salt bridges play a key role in the thermostability of this enzyme. To support their results, they constructed a salt bridge subtraction mutant and a salt bridge addition mutant by site-directed mutagenesis. The salt bridge subtraction mutant E31Q showed a 13.96% thermostability decrease, and the salt bridge addition mutant Q307D showed a 9.15% thermostability enhancement compared to the WT. In addition, Chan et al. [Citation36] used double mutant cycles to determine the temperature-dependency of the pair-wise interaction energy and the contribution of salt-bridges to △Cp in a thermophilic ribosomal protein L30e. Their results showed that the pair-wise interaction energies for the salt-bridges E6/R92 and E62/K46 were stabilizing and insensitive to temperature changes from 298 to 348 K. They suggest that the extra salt-bridges found in thermophilic proteins enhance the thermostability of proteins by reducing △Cp, leading to the up-shifting and broadening of the protein stability curves [Citation36, Citation37]. Based on a large body of research on the relationship between thermostability and salt bridges [Citation38–43], salt bridges must be closely related to protein thermostability. Higher thermostability tends to be associated with more salt bridges in proteins.
Disulphide bond prediction and mass spectrometry analysis
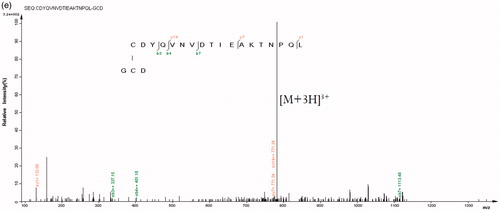
The amino acid sequence alignment between Y. NSN and S. marcescens nucleases revealed the amino acid sites which form disulphide bonds (). By using mass spectrometry, we determined the existence of disulphide bonds in Y. NSN. The HPLC LC-MS TIC spectrum of the un-reduced and the reduced sample enzymatic hydrolysate were analyzed [Citation44, Citation45]. showed that the TIC spectra of the samples were not completely consistent. The first mass spectrometry data of peptide fragments in which disulphide bonds were located were found in the database; two pairs of disulphide bonds whose matching patterns were consistent with the theory were successfully found. The XIC diagram and the mass spectrometry of disulphide bonds $1 and $2 are shown in . The secondary spectrogram of the peptide fragments in which disulphide bonds were located was verified and the linking mode of disulphide bonds $1 and $2 was confirmed ().
Figure 4. Amino acid sequence alignment of Y. NSN and non-specific nuclease from Serratia marcescens. Note: Arrows indicate the amino acid sites which form disulphide bonds.
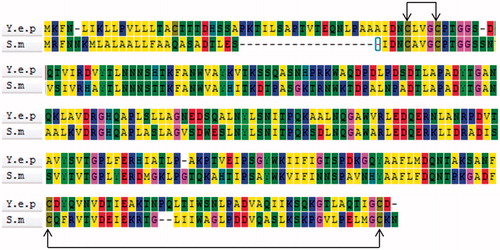
Figure 5. HPLC LC-MS spectra and diagrams of disulphide bonds of Y. NSN. (a) HPLC LC-MS TIC spectrum of enzymatic hydrolysate samples. The unreduced sample is in blue, the reduced sample is in red. (b) XIC diagram and a mass spectrometry of disulphide bond $1. (c) XIC diagram and a mass spectrometry of disulphide bond $2. (d) Secondary matching diagram of disulphide bond $1. (e) Secondary matching diagram of disulphide bond $2.
Ding et al. [Citation25] improved the thermal stability of glucose 1-dehydrogenase (LsGDH) from Lysinibacillus sphaericus G10 by introducing disulphide bridges between subunits. One out of the 11 mutants, designated as DS255, displayed significantly enhanced thermal stability with considerable soluble expression and high specific activity, having a half-life of 9900 min at 50 °0, which was 1868-fold as that of its wild type. This proved that the introduction of disulphide bonds resulted in significant improvement of the thermal stability of glucose1-dehydrogenase [Citation46, Citation47]. Moreover, in order to explore whether the formation of disulphide bonds in insect prophenoloxidases (PPOs) improved protein stability and/or increased insect innate immunity over time, Lu et al. [Citation48] used Drosophila melanogaster PPO1 as a model; one or two disulphide bonds were deleted to evaluate the importance of disulphide bonds in insect immunity. The WT PPO1 (rPPO1) and mutants lacking disulphide bonds could be expressed and showed phenoloxidase (PO) activity. The PO activities of mutants lacking one or two disulphide bonds significantly decreased and deletion of disulphide bonds also reduced the PPO thermostability.
Thermostability analysis
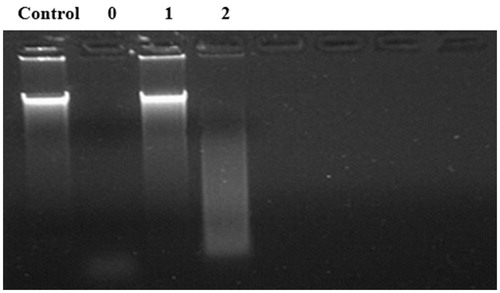
It has been widely recognized that the thermostability of proteins can be influenced by many structural factors, such as core and side-chain packing, helical content, polar surface area, hydrogen bonds and salt bridges [Citation25]. Compared to that of the WT enzyme, the salt bridge mutant R63Q was almost fully deactivated and the enzyme activity of D166Q was also significantly decreased (). Moreover, the salt bridge Arg63-Asp100 has four salt bonds. There is only one salt bond in the salt bridge Asp166-Arg169. Accordingly, R63Q showed lower thermostability than D166Q. These results support the idea that the thermostability of Y. NSN was proportional to the amount of salt bonds and that salt bridges could play an important role in the thermostability of this enzyme. These results are in agreement with Fei et al. [Citation26], who studied the relationship between the thermostability of phytase and its salt bridges and reported similar results. Thus, salt bonds should be a key factor with higher priority for consideration to enhance the thermostability of Y. NSN.
Figure 6. Effect of salt bridges on the thermostability of Y. NSN. Control: no nuclease; Lane 0: WT; Lane 1: R63Q; Lane 2: D166Q.
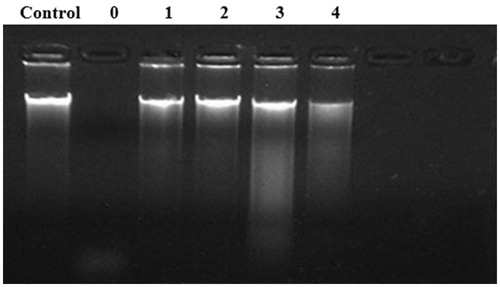
Disulphide bonds play a vital role in stabilizing the protein spacial structure [Citation48, Citation49]. The essential role of the two disulphide bonds in the activity of S. marcescens nuclease has been demonstrated by site-directed mutagenesis [Citation26]. We also found high similarity in the sequence of Y. NSN and S. marcescens nuclease. The mode of connection and pairings of the disulphide bonds in Y. NSN were confirmed through mass spectrum identification. There are two pairs of disulphide bonds in S. marcescens nuclease; thus, we speculated that Y. NSN similarly forms two pairs of disulphide bonds at the same amino acid sites, which are C18-C22 and C208-C252. Therefore, the cysteine engaged in disulphide bond formation in Y. NSN was mutated. After incubation at 80 °0 for 30 min, compared to that of the WT enzyme, the activities of the mutants C18S, C22S, C208S and C252S were nearly completely inactivated, and the mutant C208S only retained very low enzyme activity (). The reason for all the four mutants being almost fully deactivated might be due to the rupture of the disulphide bonds, which changed the spacial structure of Y. NSN and then affected its thermo stability. These results verified that disulphide bonds were another factor that influenced the thermostability of Y. NSN.
Figure 7. Effect of disulphide bonds on the thermostability of Y. NSN. Control: no nuclease; Lane 0: WT; Lane 1: C18S; Lane 2: C22S; Lane 3: C208S; Lane 4: C252S.
Conclusions
Based on the comprehensive analysis of the spatial structure of Y. NSN, by using site-directed mutagenesis, we demonstrated that changes in the active site played an important role in the enzyme activity of Y. NSN. Salt bridges were a key factor affecting the thermo stability of Y. NSN and the number of salt bonds was proportional to its thermo stability. In addition, disulphide bonds are another factor that plays an important part in the thermo stability of Y. NSN. Therefore, manipulation of the salt bonds and disulphide bonds could be used for enhancement of the thermostability of Y. NSN.
Disclosure statement
No potential conflict of interests was reported by the author(s).
Additional information
Funding
References
- Hsia KC, Li CL, Yuan HS. Structural and functional insight into sugar-nonspecific nucleases in host defense. Curr Opin Struct Biol. 2005;15:126–134.
- Meiss G, Friedhoff PM, Gimadutdinow O, et al. Sequence preferences in cleavage of dsDNA and ssDNA by the extracellular Serratia marcescens endonuclease. Biochemistry. 1995;34:11979–11988.
- Yi-Ting W, Wei-Jen Y, Chia-Lung L, et al. Structural basis for sequence-dependent DNA cleavage by nonspecific endonucleases. Nucleic Acids Res. 2007;35:584–594.
- Kandavelou K, Mani M, Durai S, et al. Engineering and applications of chimeric nucleases. Nucleic Acids Mol Biol. 2004;14:413–434.
- Li L, Lin S, Feng Y. Functional identification of the non-specific nuclease from white spot syndrome virus. Virology. 2005;337:399–406.
- Maclellan SR, Forsberg CW. Properties of the major non-specific endonuclease from the strict anaerobe Fibrobacter succinogenes and evidence for disulfide bond formation in vivo. Microbiology. 2001;147:315–323.
- Samejima K, Earnshaw WC. Trashing the genome: the role of nucleases during apoptosis. Nat Rev Mol Cell Biol. 2005;6:677–688.
- Falcone RJ, Stern L, Shin C, et al. Apoptosis and the pattern of DNase I expression following massive small bowel resection. J Surg Res. 1999;84:218–222.
- Fang XJ, Tang ZX, Li ZH, et al. Production of a new non-specific nuclease from Yersinia enterocolitica subsp. palearctica: optimization of induction conditions using response surface methodology. Biotechnol Biotec Equip. 2014;28:559–566.
- Yu Z, Zhen-Hua L, Wei Z, et al. Enzyme activity and thermostability of a non-specific nuclease from Yersinia enterocolitica subsp. palearctica by site-directed mutagenesis. Electron J Biotech. 2016;24:32–37.
- Hejazi A, Falkiner FR. Serratia marcescens. J Med Microbiol. 1997;46:903–912.
- Li X, Tetling S, Winkler UK, et al. Gene cloning, sequence analysis, purification, and secretion by Escherichia coli of an extracellular lipase from Serratia marcescens. Appl Environ Microb. 1995;61:2674–2680.
- Linbo L, Manickam K, Baseman JB, et al. Molecular cloning, expression, and characterization of a Ca2+-dependent, membrane-associated nuclease of Mycoplasma genitalium. J Bacteriol. 2010;192:4876–4884.
- Oliveri M, Daga A, Lunardi C, et al. DNase I behaves as a transcription factor which modulates Fas expression in human cells. Eur J Immunol. 2004;34:273–279.
- Panfilova ZI, Salganik RI. Isolation of Serratia marcescens mutants superproducers of endonuclease by exposure to nitrosomethylurea in a synchronized culture. Mikrobiologiia. 1983;52:974–978.
- Song Q, Zhang X. Characterization of a novel non-specific nuclease from thermophilic bacteriophage GBSV1. BMC Biotechnol. 2008;8:43.
- Benedik MJ, Strych U. Serratia marcescens and its extracellular nuclease. FEMS Microbiol Lett. 1998;165:1–13.
- Mahua G, Gregor M, Alfred P, et al. Structural insights into the mechanism of nuclease A, a betabeta alpha metal nuclease from Anabaena. J Biol Chem. 2005;280:27990–27997.
- Cinatl J, Anand P, Rothweiler F. Onconase induces caspase-independent cell death in chemoresistant neuroblastoma cells. Cancer Lett. 2007;250:107–116.
- Gast FU, Franke I, Meiss G, et al. Immobilization of sugar-non-specific nucleases by utilizing the streptavidin-biotin interaction. J Biotechnol. 2001;87:131–141.
- Melvin MS, Calcutt MW, Noftle RE, et al. Influence of the a-ring on the redox and nuclease properties of the prodigiosins: importance of the bipyrrole moiety in oxidative DNA cleavage. Chem Res Toxicol. 2002;15:742–748.
- Ohkura T, Yamada K, Okamoto A, et al. Nationwide epidemiological study revealed the dissemination of meticillin-resistant Staphylococcus aureus carrying a specific set of virulence-associated genes in Japanese hospitals. J Med Microbiol. 2009;58:1329–1336.
- Schulthess B, Bloes DA, Francois P, et al. The σB-dependent yabJ-spoVG operon is involved in the regulation of extracellular nuclease, lipase, and protease expression in Staphylococcus aureus. J Bacteriol. 2011;193:4954–4962.
- Li ZH, Tang ZX, Fang XJ, et al. Bioinformatics analysis of a non-specific nuclease from Yersinia enterocolitica subsp. palearctica. Comput Biol Chem. 2013;47:207–214.
- Ding H, Gao F, Liu D, et al. Significant improvement of thermal stability of glucose 1-dehydrogenase by introducing disulfide bonds at the tetramer interface. Enzyme Microb Tech. 2013;53:365–372.
- Fei B, Hui X, Zhang F, et al. Relationship between Escherichia coli AppA phytase’s thermostability and salt bridges. J Biosci Bioeng. 2013;115:623–627.
- Beek HLV, Wijma HJ, Fromont L, et al. Stabilization of cyclohexanone monooxygenase by a computationally designed disulfide bond spanning only one residue. Febs Open Bio. 2014;4:168–174.
- Friedhoff P, Gimadutdinow O, Pingound A. Identification of catalytically relevant amino acids of the extracellular Serratia marcescens endonuclease by alignment-guided mutagenesis. Nucleic Acids Res. 1994;22:3280–3287.
- Matak MY, Noghaddam ME. The role of short-range Cys171-Cys178 disulfide bond in maintaining cutinase active site integrity: a molecular dynamics simulation. Biochem Bioph Res Commun. 2009;390:201–204.
- Fisher CL, Pei GK. Modification of a PCR-based site-directed mutagenesis method. Biotechniques. 1997;23:570–574.
- Gao F. Structural and functional study of Agrobacterium nuclease Atu4108 and Salmonella typhimuriun regulators STM1987 and Art [master’s thesis]. Jinan (SD): Shan Dong University; 2017.
- Qing L, Chiu NHL, Chang S, et al. Investigation of enzymatic behavior of benzonase/alkaline phosphatase in the digestion of oligonucleotides and DNA by ESI-LC/MS. Anal Chem. 2007;79:1907–1917.
- Zhou JY, Petritis BO, Petritis K, et al. Mouse-specific Tandem IgY7-supermix immunoaffinity separations for improved LC-MS/MS coverage of the plasma proteome. J Proteome Res. 2009;8:5387–5395.
- Bessler C, Schmitt J, Maurer KH, et al. Directed evolution of a bacterial alpha-amylase: toward enhanced pH-performance and higher specific activity. Protein Sci. 2003;12:2141–2149.
- Jing G, Rao Z, Yang T, et al. Enhancement of the thermostability of Streptomyces kathirae SC-1 tyrosinase by rational design and empirical mutation. Enzyme Microb Tech. 2015;77:54–60.
- Chi-Ho C, Tsz-Ha Y, Kam-Bo W. Stabilizing salt-bridge enhances protein thermostability by reducing the heat capacity change of unfolding. Plos One. 2011;6:e21624.
- Sabiani S, Geppert T, Engelbrecht C, et al. Unraveling the activation mechanism of Taspase1 which controls the oncogenic AF4-MLL fusion protein. Ebiomedicine. 2015;2:386–395.
- Bikkina S, Bhati A P, Padhi S, et al. Temperature dependence of the stability of ion pair interactions, and its implications on the thermostability of proteins from thermophiles. J Chem Sci. 2017;129:1–10.
- Chen X, Liu X, Fang Y. Memeli prion proteinlerinin moleküler dinamik simulasyon ile tuz köprüleri etkilesimlerinin arastirilmasi. [Investigation on salt bridge interactions of mammalian prion proteins by molecular dynamics simulation.] Turkish J Biochem. 2016;41:177–188.
- Kuhn I, Kellenberger E, Cakir-Kiefer C, et al. Probing the catalytic mechanism of bovine CD38/NAD + glycohydrolase by site directed mutagenesis of key active site residues. Biochim Biophys Acta. 2014;1844:1317–1331.
- Liao T. Deoxythymidine 3', 5'-di-p-nitrophenyl phosphate as a synthetic substrate for bovine pancreatic deoxyribonuclease. J Biol Chem. 1975;250:3721–3724.
- Folch B, Rooman M, Dehouck Y. Thermostability of salt bridges versus hydrophobic interactions in proteins probed by statistical potentials. J Chem Inf Model. 2008;48:119–127.
- Fu Y, Ding Y, Wang Z, et al. Study on the relationship between cyclodextrin glycosyltransferase thermostability and salt bridge formation by molecular dynamics simulation. Protein Peptide Lett. 2010;17:1403–1411.
- Ge M, Xia XY, Pan XM. Salt bridges in the hyperthermophilic protein Ssh10b are resilient to temperature increases. J Biol Chem. 2008;283:31690–31696.
- Missimer JH, Steinmetz MO, Baron R, et al. Configurational entropy elucidates the role of salt‐bridge networks in protein thermostability. Protein Sci. 2007;16:1349–1359.
- Dominy BN, Minoux H, Charles LB. An electrostatic basis for the stability of thermophilic proteins. Proteins. 2004;57:128–141.
- Tuhin G, Shekhar G, Garcia AE. Role of backbone hydration and salt-bridge formation in stability of alpha-helix in solution. Biophys J. 2003;85:3187–3193.
- Lu A, Peng Q, Ling E. Formation of disulfide bonds in insect prophenoloxidase enhances immunity through improving enzyme activity and stability. Dev Comp Immunol. 2014;44:351–358.
- Pilipczuk J, Zalewska-Piątek B, Bruździak P, et al. Role of the disulfide bond in stabilizing and folding of the fimbrial protein DraE from uropathogenic Escherichia coli. J Bio Chem. 2017;292:16136–16149.