
Abstract
Neural stem cells (NSCs) have the ability to differentiate into neurons, astrocytes and oligodendrocytes. They are self-renewing and sufficient to provide large amounts of brain tissue cells. NSCs have promising application prospects for the treatment of central nervous system diseases. Spider toxins are important tools for use in neurobiology and neuropharmacology. In this study, a Cell Counting Kit (CCK-8), immunofluorescence staining, real-time quantitative polymerase chain reaction (RT-qPCR) and Western blotting were used to qualitatively and quantitatively analyse the effects of different concentrations (1, 10 and 20 μg/mL) of Chilobrachys jingzhao crude venom on the proliferation and differentiation of C17.2 cells in vitro. At low concentrations, C. jingzhao crude venom did not significantly inhibit the proliferation of NSCs; however, the highest concentration of 20 μg/mL partially inhibited the NSC proliferation. In the differentiation medium, addition of C. jingzhao crude venom effectively promoted the differentiation of NSCs towards neuronal cells, with a final concentration of 10 μg/mL having the most obvious effect. These results suggest that C. jingzhao crude venom exerts a regulatory effect on the proliferation and differentiation of C17.2 cells. Therefore, spider venom can be used as an ‘ammunition library’ to find factors that regulate the proliferation and differentiation of NSCs, laying the groundwork for the application of NSCs in the treatment of central nervous system diseases.
Introduction
According to fossil records, spiders are an ancient species with a history spanning 400 million years, more than 150 million years earlier than even the dinosaurs [Citation1–3]. For predation and defence against enemies, the spider venom gland gradually evolved into a complex, rich ‘treasure trove’ that contains abundant toxins targeting a wide range of receptors, channels and enzymes of vertebrates and invertebrates [Citation4].
The components of the spider's crude venom belong to the following three categories: small-molecular-weight toxins (molecular weight <1000 Da), low-molecular-weight toxins (molecular weight between 1000 Da and 10 kDa) and high-molecular-weight toxins (molecular weight >10 kDa). The small-molecular-weight material’s core components include inorganic salts, free acids, free amino acids, biogenic amines, neurotransmitters and acyl polyamines. The polyamines in spider toxins are neurotoxic and act on ionotropic glutamate receptors [Citation5], voltage-dependent open channels [Citation6], voltage-sensitive calcium channels [Citation7] and acetylcholine (ACh) receptors. In addition, these polyamines act as antimicrobial substances [Citation8]. Most of the low-molecular-weight toxins can interact with various ion channels such as Nav, Kv and Cav. The high-molecular-weight toxins are primarily derived from the black widow spider. For example, α-latrotoxin’s relative molecular weight is above 100 × 103, and it can bind presynaptic membranes, causing neuroticism [Citation9].
Since spiders employ their venom primarily to paralyze their predators or prey, the venom contains a large number of peptides that regulate neuronal ion channels and receptor activity. Some toxins whose structure and mechanism of action have been validated have become irreplaceable tools for research in neurobiology and neuropharmacology with respect to ion channels, neurotransmitter receptors, synaptic transmission and so on. In addition, the high target affinity and selectivity of the toxins for ion channels also make spider venom peptides an ideal natural source for the discovery of novel therapeutic targets [Citation10].
The Chinese tarantula, Chilobrachys jingzhao (syn. Chilobrachys guangxiensis), is one of the most venomous spiders inhabiting the hilly areas of Hainan province in China, and it belongs to the genus of huwena (Theraphosidae), rod thorn spider (Selenocosmiinae), and tassel hair spider (Chilobrachys). C. jingzhao’s venom contains a variety of natural neurotoxins. For example, JZTX-I preferentially inhibits the activation of sodium receptors and interacts with channel isoforms expressed in mammalian and insect sensory neurons [Citation11]. JZTX-V inhibits both tetrodotoxin-resistant and tetrodotoxin-sensitive sodium currents in rat dorsal root ganglion neurons [Citation12]. The research on the toxicity of C. jingzhao crude venom on the nervous system is not new [Citation13]. C. jingzhao crude venom inhibits the excitatory transmission in the sciatic nerve of toad muscle brought about by electrical stimulation. In the low concentration range, C. jingzhao crude venom produces a concentration-dependent inhibitory effect on muscle contraction induced by nerve stimulation, but it has no impact on the nerve action potential (NAP). High concentrations of spider venom have an irreversible inhibitory effect on muscle contractions induced by direct stimulation, which damages the muscle fibres and causes muscle spasms.
Central nervous system degenerative diseases exhibit pathologically visible brain and/or spinal neuron degeneration and loss. Major diseases include Parkinson's disease (PD), Alzheimer's disease (AD), Huntington disease (HD) and amyotrophic lateral sclerosis (ALS). These diseases represent some of the most difficult to treat among neurological diseases. Traditional methods include drug treatments and surgical treatments, the former of which can relieve symptoms but cannot prevent progression of the disease. The latter is not an ideal treatment due to the risk for inducing trauma. Exploring new strategies for the treatment of neurological diseases has become a hot topic in current research trends. Neural transplantation using donor human embryonic nerve tissue has provided an effective approach for the treatment of PD and AD [Citation14]. However, limited donor sources prevent the widespread use of this approach. The use of stem cells can potentially overcome these problems. First, neural stem cells (NSCs) can be expanded in vitro, and second, through in vitro expansion of the patient's own stem cells, they can be re-implanted into the brain, avoiding potential immune rejection. Therefore, basic and clinical research on the use of NSC transplantation in the treatment of central nervous system degeneration has generated much interest [Citation15, Citation16]. If NSCs are successfully cultured and induced to differentiate into a specific phenotype, they can provide a rich source of cells for experimental studies and neural transplants in the treatment of degenerative brain diseases.
The addition of neurotrophic and growth factors to culture medium is one of the most precise methods to control the proliferation and differentiation of NSCs. At present, brain-derived neurotrophic factor (BDNF) [Citation17], neurotrophin-3 (NT-3) [Citation18], nerve growth factor (NGF) [Citation19] and epidermal growth factor (EGF) [Citation20] are the most widely used. BDNF acts by triggering the Wnt/β-catenin signalling pathway in NSCs [Citation21]. BDNF stimulates the proliferation of NSCs and significantly increases their differentiation into neurons and oligodendrocytes. BDNF has been used to treat AD [Citation22]. Studies using spider toxins for application in the treatment of AD and other central nervous system degenerative diseases have also been reported [Citation23, Citation24]. Although spider toxins have great potential for the treatment of central nervous system degenerative diseases, there are scarcely any data describing the interaction of spider venom with NSCs or the effect of spider toxins on the differentiation and proliferation of NSCs.
In this study, we examined the effects of varying concentrations of C. jingzhao crude venom (1, 10 and 20 μg/mL, with fresh medium without venom as a control) on the proliferation and differentiation of NSCs using CCK-8, immunofluorescence staining, real-time reverse transcription polymerase chain reaction (RT-qPCR) and Western blot. C17.2 cells were used to detect the proliferation and differentiation of NSCs. C17.2 cells are an immortalised mouse neural progenitor cell line capable of differentiation in vitro. The cell line was originally established using retroviral-mediated transduction of the avian myc oncogene into mitotic progenitor cells from a neonatal mouse cerebellum, which is a valuable tool for in vitro and in vivo studies into the control of cell fate and differentiation of neural progenitors [Citation25–27].
Materials and methods
Materials
Chilobrachys Jingzhao crude venom was obtained from the Key Laboratory of Protein Chemistry and Developmental Biology of the Ministry of Education, College of Life Sciences, Hunan Normal University. C17.2 NSCs were a generous gift from the Key Laboratory of Protein Chemistry and Developmental Biology of the Ministry of Education, College of Life Sciences, Hunan Normal University. Dulbecco's modified Eagle medium containing nutrient mixture F-12 (DMEM/F-12), horse serum, fetal bovine serum (FBS), phosphate-buffered saline (PBS), 10,000 U/mL penicillin and 10,000 μg/mL streptomycin were purchased from Gibco (USA). Trypsin solution (0.25%) was purchased from HyClone (USA). Recombinant mouse β-NGF (N1408) and recombinant human BDNF (B3795) were purchased from Sigma (USA). 4,6-Diamidino-2-phenylindole (DAPI) and Cell Counting Kit-8 (CCK-8) were purchased from Dojindo (Japan). SYBR Green Master Mix and Trizol reagents were purchased from Life technologies (USA). All primers for the real-time reverse transcription quantitative polymerase chain reaction (RT-qPCR) were purchased from Sangon (Shanghai, China). Antibodies against glial fibrillary acidic protein (GFAP) (ab7260), β-tubulin III (ab7751) and MBP2 for Western blot and immunofluorescence were purchased from Abcam (UK). An antibody against β-actin (sc-1616) was purchased from Beyotime (China). Horseradish peroxidase (HRP) AffiniPure Goat Anti-Mouse IgG (E030110), HRP Affinipure Goat Anti-Rabbit IgG (E030120) and HRP AffiniPure Rabbit Anti-Chicken IgG were purchased from Earthox (USA). Fluorescein isothiocyanate (FITC)-conjugated AffiniPure Goat Anti-Rabbit IgG (BA1105) and FITC-conjugated AffiniPure Goat Anti-Mouse IgG (BA1101) were purchased from Boster (China); FITC-conjugated AffiniPure Donkey Anti-Chicken IgG (D110202) was purchased from BBI (China). All plasticware used for cell culturing was purchased from Corning Inc. (USA).
Proliferation cultures of C17.2 cells
For conventional cultures, C17.2 cells were seeded in cell proliferation medium at a density of 1.0 × 103 cells/mL. The proliferation culture medium mainly consisted of DMEM/F-12 medium supplemented with 5% horse serum, 10% FBS, 100 U penicillin/mL and 100 μg streptomycin/mL. The confluent cells were detached every third day using 0.25% trypsin solution and seeded in a new cell culture dish at a ratio of one to three.
Differentiation cultures of C17.2 cells
For differentiation studies, C17.2 cells were seeded in proliferation medium (according to the needs of individual experiments, inoculated at the corresponding density). About 24 h after seeding, the medium was changed to differentiation medium, comprising DMEM/F-12 medium, 100 U penicillin/mL, 100 μg streptomycin/mL, modified N2 supplements (to a final concentration of 5 μg/mL bovine insulin, 20 nmol/L progesterone, 30 nmol/L sodium selenite, 100 μg/mL bovine apo-transferrin and 100 μmol/L putrescine dihydrochloride), 10 ng/mL NGF and 10 ng/mL BDNF. Differentiation medium was changed every third day for the duration of differentiation.
C. jingzhao crude venom treatment
Chilobrachys jingzhao crude venom was dissolved in PBS to a concentration of 1 mg/mL. The stock solution was filtered through a 0.2-μm sterile filter and added to the proliferation culture medium and the differentiation culture medium, diluted to the final concentrations of 1, 10 and 20 μg/mL, or fresh medium without venom for the control group experiments.
Proliferation analysis of C17.2 cells
Cell proliferation experiments were performed in 96-well plates. Cells were seeded at a density of 1 × 103 cells/well and cultured in the proliferation culture medium, followed by exposure to 1, 10 or 20 μg/mL C. jingzhao crude venom. After 1, 3 and 5 days, 10 μL of CCK-8 solution was added to each well. After incubation at 37 °C for 4 h, the absorbance of WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt] in each well was measured at 450 nm with a Microplate reader (MULTISKAN FC, Thermo, USA).
Immunofluorescence staining
C17.2 cells were seeded in 6-cm-diameter culture dishes at a density of 1.3 × 104 cells/mL. The expression levels of β-tubulin III, GFAP and MBP2 were used to characterise the differentiation of C17.2 cells using experimental methods similar to those described previously [Citation28]. After culturing cells in differentiation medium for 10 days, the medium was removed, the cells were rinsed with PBS, fixed with paraformaldehyde for 30 min at room temperature, and incubated in PBS with 0.1% Triton X-100 (PBST) for 30 min to allow cells to permeabilise. Then, the cells were blocked using normal goat serum for 1 h at room temperature followed by incubation at room temperature for 2 h in PBST containing diluted primary antibodies (1:500 for β-tubulin III and 1:1000 for GFAP and MBP2). After washing, the cells were incubated for 1 h in PBST containing secondary antibodies conjugated to FITC (1:1000). Nuclear staining was performed by incubating the cells in a solution of PBST with DAPI (1:1000) for 5 min. Finally, the cells were washed with PBST three times, and fluorescence was visualised with a fluorescence microscope (Leica, DMI4000B, Germany).
RT-qPCR
C17.2 cells were seeded in 6-cm-diameter culture dishes at a density of 6.7 × 104 cells/mL. Cells were harvested after 10 days in differentiation media and lysed using Trizol reagent following incubation at room temperature for 5 min. After shaking with chloroform, cells were incubated at room temperature for 3 min and lysates were centrifuged at 12,000×g for 15 min at 4 °C. After centrifugation, the solution was divided into three layers. The upper aqueous phase containing the total RNA was placed into a new tube. Isopropanol was added to the aqueous phase and incubated at room temperature for 10 min. RNA was precipitated by centrifugation at 12,000×g for 10 min at 4 °C. Total RNA was washed with 75% ethanol, allowed to air dry, and dissolved in RNase-free water. For each sample, 2 μg RNA was transcribed into cDNA. Oligo dTs, dNTPs and total RNA were added together. After incubation at 65 °C for 5 min, the tube was chilled on ice for 1 min. After adding RT buffer, magnesium chloride (MgCl2), dithiothreitol (DTT), RNase inhibitor and SuperScript III, the tube was incubated at 50 °C for 50 min following incubation at 85 °C for 5 min to obtain cDNA. cDNA was subsequently used for RT-qPCR using SYBR green with real-time fluorescence quantitative PCR instrument (LightCycler 480II, Roche, Switzerland). A list of gene-specific primers is shown in . Amplifications (2720 Thermal Cycler, Applied Biosystems, USA) were performed with predenaturation at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 15 s, annealing at 55 °C for 1 min, and elongation at 60 °C for 1 min. The PCR products were analysed on 1.5% agarose gels and visualised with ethidium bromide and UV radiation.
Table 1. RT-qPCR primer sequences.
Western blot
C17.2 cells were seeded in 9-cm-diameter culture dishes at a density of 1.3 × 105 cells/mL. After 10 days in differentiation media, cells were harvested. Cells were washed three times with ice-cold PBS and lysed with TEN-T buffer (150 mmol/L NaCl, 10 mmol/L Tris-HCl, pH 7.4, 5 mmol/L EDTA, pH 8.0, 1% Triton X-100, 1 mmol/L PMSF and 2 µg/mL of aprotinin), and 60 µg total protein lysate (concentrations determined by the Bradford assay according to the manufacturer’s instructions) were separated in 10% sodium dodecyl sulphate (SDS)-polyacrylamide gels. Proteins were transferred to polyvinylidene fluoride (PVDF) membranes for blotting. The membranes were blocked with 5% skim milk for 2 h at room temperature, and blotted overnight at 4 °C with the mouse anti-β-tubulin III (1:10,000), rabbit anti-GFAP (1:10,000) and chicken anti-MBP2 (1:10,000) primary antibodies. HRP-conjugated secondary antibodies anti-mouse IgG (1:4000), anti-rabbit IgG (1:4000) and anti-chicken IgG (1:4000) were applied. Mouse anti-β-actin (1:1000) was used as a loading control followed by HRP-conjugated anti-mouse IgG (1:4000) secondary antibody. A blot scanner (C-DiGit®, LI-COR, USA) was used to detect the band density of the proteins of interest. The results were analysed using ImageJ software.
Statistical analysis
Data are expressed as mean values with standard deviation (±SD) from at least three independent experiments. Statistical comparisons were performed using analysis of variance (ANOVA), and differences at P < 0.05 were considered statistically significant.
Results and discussion
The effect of different concentrations of venom on C17.2 cell proliferation
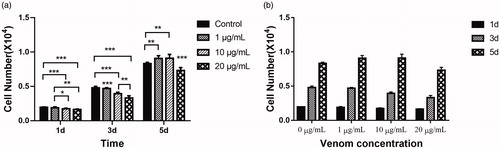
shows C17.2 cell proliferation from days 1 to 5. In general, the number of cells increased with time (). Specifically, on the first day (), the cells in the control group without crude venom exhibited the fastest growth (the number of cells increased by 95.33%), followed by the experimental group treated with 10 μg/mL toxin (the number of cells increased by 88.71%). The experimental groups treated with 10 or 20 μg/mL venom showed relatively low rates of cell proliferation, with cell numbers increasing by 70.08% and 63.35%, respectively. By the third day, the differences in cell numbers between the groups were more pronounced, showing an inverse relationship between venom concentration and proliferation rates. In the controls, the number of cells increased by 4.78-fold from the number of cells inoculated, whereas the number of cells treated with a crude venom concentration of 10 μg/mL increased by only 3.92-fold from the initial cell count. The number of cells treated with crude venom at a concentration of 20 μg/mL increased by a mere 2.31-fold from the initial number of cells seeded. By the fifth day, the proliferation of C17.2 cells in the 1 and 10 μg/mL treated groups had improved, exhibiting 806.83% and 810.16% increases, respectively, compared with the cell count at the time of inoculation, whereas the number of cells in the control group had increased by 732.83%. The cells in the 20 μg/mL crude venom treated group showed the smallest increase at 631.83%. Based on the above results, in the proliferation medium, we observed over a short time (the first day, the third day) that C. jingzhao venom inhibited the proliferation, with the most pronounced inhibition occurring in the group with the two higher venom concentrations. With increasing culture time, this inhibitory effect weakened, and the cell proliferation rates improved. The results can be explained as follows: When the concentrations of crude venom were relatively low, negative effects were exerted on cell proliferation (e.g. cell death), but with increasing time in cell culture, the cells proliferated rapidly enough to eventually compensate for the negative effects from toxins. In contrast to previously reported neurotoxin compounds, such as acrylamide, a compound that significantly inhibits NSC proliferation [Citation27], or ketamine, an anaesthetic that alters neurogenesis and regulates NSC differentiation by inhibiting the proliferation of NSCs [Citation29], the most significant effect of C. jingzhao crude venom is the inhibitory effect cell proliferation that can be overcome at low doses. The non-inhibitory effect of low concentrations of crude poisons on the proliferation of NSCs is undoubtedly beneficial for the research and development of the technology of stem cell transplantation. After all, as a powerful tool for the treatment of central nervous system diseases, the most important thing is to ensure that the proliferative activity of the transplanted stem cells can be normalised so as to obtain sufficient therapeutic raw materials, i.e. NSCs. It is worth mentioning that when the concentrations of venom reached 20 μg/mL, the negative effects were enhanced, and cell proliferation was unable to completely recover to control levels. In particular, it should be noted that when the toxin concentration is further increased to 50 or 100 μg/mL, C17.2 cells are unable to survive (data not shown).
Figure 1. Cell proliferation analysis on days 1, 3 and 5 of neural progenitor C17.2 cells cultured in proliferation culture medium with three different venom concentrations. Effect of crude venom concentration (a) and incubation time (b).
Note: Mean values; error bars represent ± SD (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001.
Morphology of differentiated C17.2 cells
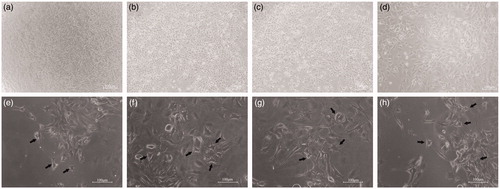
shows the morphology of C17.2 cells cultured for 6 days in either the proliferation medium or the differentiation medium and various concentrations of crude venom. Observations under the microscope showed that the number of C17.2 cells cultured in proliferation medium increased rapidly, with retention of native and NSC morphology. Morphological characteristics of neurons were not observed in the cells grown in the proliferation medium (). In contrast, the cells cultured for 6 days in the differentiation medium clearly adopted another form, which was easily distinguished from the cells that maintained the native C17.2 cell morphology. The morphological characteristics of these cells included extension of elongated synapses, indicating neuronal cell characteristics (). It is worth mentioning that we observed greater development of neuronal-like cells cultured in differentiation medium with the addition of crude venom. These phenomena suggest that there is a possibility that the addition of crude venom in the differentiation medium may favour the differentiation of NSCs towards neurons. This hypothesis needs to be further verified by subsequent experiments.
Figure 2. Morphology of neural progenitor C17.2 cells cultured in proliferation medium (a–d) or differentiation medium (e–h) for 6 days. Control cells (a), cells exposed to 1 μg/mL (b), cells exposed to 10 μg/mL (c) and cells exposed to 20 μg/mL (d) of crude venom dissolved in the proliferation medium. Control cells (e), cells exposed to 1 μg/mL (f), cells exposed to 10 μg/mL (g) and cells exposed to 20 μg/mL (h) of crude venom dissolved in differentiation medium.
Note: Phase contrast images. Scale bar 100 μm.
Immunostaining of cell type-specific biomarkers in differentiated C17.2 cells
To verify the cell changes in response to the differentiation medium, determine the direction of cell differentiation, and understand the effects of C. jingzhao crude venom on cell differentiation, we used immunofluorescence staining, RT-qPCR and Western blotting to qualitatively and quantitatively analyse the cell type-specific biomarkers during the C17.2 cell differentiation.
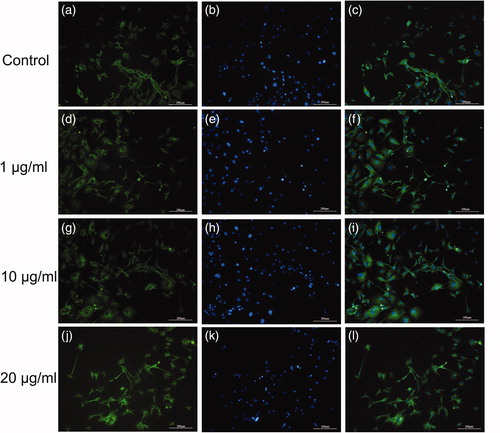
For immunofluorescence detection, GFAP (an astrocyte marker) and β-tubulin III (an early neuronal marker) were used after 10 days in differentiation medium and C. jingzhao venom. shows that the majority of cells displayed positive immunoreactivity for β-tubulin III when cultured in differentiation medium, but almost no GFAP expression was seen (data not shown). With respect to any concentration-dependent effects of the crude venom on the cell differentiation, the number of β-tubulin III-positive cells in the medium with crude venom was higher than that in the medium without crude venom. In addition, more β-tubulin III-positive cells were observed in the differentiation medium containing a venom concentration of 1 and 10 μg/mL. As shown in , neuronal-like cells were observed under the optical microscope () identified by positive β-tubulin III immunoreactivity. This result suggests that the C17.2 NSCs cultured in differentiation medium preferentially differentiate into neurons, and addition of crude venom into the culture medium facilitates this process.
Figure 3. Effects of venom on differentiation of neural progenitor cells. β-Tubulin III (green) and nuclei (blue) in control cells (a–c), cells exposed to 1 μg/mL (d–f), cells exposed to 10 μg/mL (g–i) and cells exposed to 20 μg/mL (j–l) of crude venom. Note: Immunofluorescence images. Scale bars: 200 μm. C17.2 cells were exposed to either 1, 10 or 20 μg/mL of C. jingzhao crude venom in differentiation medium for 10 days.
mRNA and protein levels of cell type-specific biomarkers in differentiated C17.2 cells
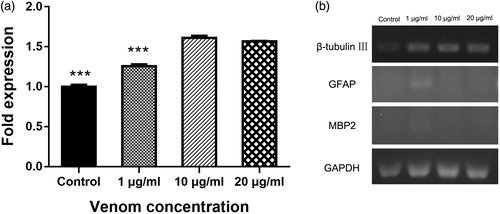
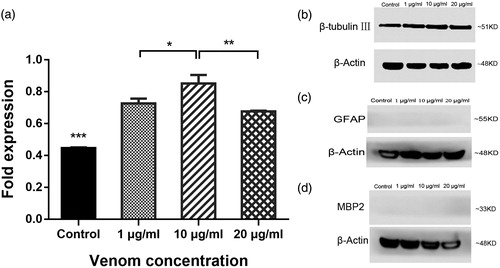
To comprehensively and objectively evaluate the effects of venom on C17.2 cell differentiation, we measured the expression levels of mRNA and protein biomarkers that are significant for differentiation. After 10 days of culture in differentiation medium in the presence of venom, we verified the differentiation capacity of C17.2 cells by assessing the expression of GFAP, β-tubulin III and MBP2 (an oligodendrocyte marker) using RT-qPCR. Consistent with the morphology observations and immunofluorescence staining results, C17.2 cells cultured in differentiation medium exhibited almost no expression of GFAP or MBP2 by RT-qPCR (). The mRNA levels of GFAP could be detected after 35 PCR cycles, while the mRNA levels of MBP2 were undetectable even after 40 PCR cycles. Likewise, PCR amplification of GFAP and MPB2 was also undetected by agarose gel electrophoresis. Therefore, we focused on the effects of the crude venom on neuronal cell differentiation. Cells cultured in differentiation medium with a venom concentration of 10 μg/mL exhibited the highest expression of β-tubulin III, indicating that this concentration of crude venom most potently promoted neuronal differentiation ().
Figure 4. Effects of venom on expression levels of neuronal biomarkers in vitro. mRNA levels of β-tubulin III after 10 days differentiation and crude venom exposure (a). Specific gene expression determined by RT-PCR (b). Note: C17.2 cells were exposed to 1, 10 or 20 μg/mL of C. jingzhao crude venom in differentiation medium for 10 days. Error bars represent means ± SD (n = 3). ***P < 0.001.
Concomitant with mRNA expression, the protein levels of GFAP, β-tubulin III and MBP2 were determined by Western blot analysis after culturing cells for 10 days in differentiation medium with venom. Total GFAP and MBP2 protein levels were not easy to detect (). Additionally, in agreement with the mRNA expression results, the cells cultured in differentiation medium with 10 μg/mL venom exhibited the highest expression of β-tubulin III (), i.e. C17.2 cells exposed to 10 μg/mL C. jingzhao crude venom were more inclined to differentiate into neurons compared to the other venom concentrations.
Figure 5. Effects of C. jingzhao crude venom on expression levels of neuronal biomarkers in vitro. Protein levels of β-tubulin III in response to differentiation and crude venom exposure (a). Representative Western blot staining for β-tubulin III (b), GFAP (c), MBP2 (d) and the loading control β-actin. Note: C17.2 cells were exposed to 1, 10 or 20 μg/mL of venom during 10 days of differentiation. Error bars represent means ± SD (n = 3) *P < 0.05, **P < 0.01, ***P < 0.001.
These results consistently demonstrated that C17.2 cells cultured in differentiation medium with a venom concentration of 10 μg/mL exhibited increased neuronal differentiation. As mentioned earlier, NSC transplantation in the treatment of these diseases is an application with great value. Studies have shown that it is not ideal to simply transplant NSCs because implanted cells are primarily transformed into astrocytes [Citation30]. Therefore, clarifying the factors that regulate the proliferation and differentiation of NSCs is key to the successful use of NSCs in the treatment of central nervous system injury. Identifying factors that can influence the differentiation of NSCs to neurons has become a hot topic for many researchers [Citation31–35]. The effects of neurotrophic factors, NGFs and cell cycle-associated proteins on the proliferation and differentiation of NSCs have been reported in detail. For instance, BDNF stimulates the proliferation of NSCs and significantly increases their differentiation into neurons and oligodendrocytes [Citation21]. NT-3 promotes the proliferation and differentiation of BM-NSCs into cholinergic neurons and increases the levels of ACh [Citation36]. NGF has the dual biological function of providing neuronal nutrition and promoting the growth of protrusions [Citation31]. The neural system-specific transmembrane protein LINGO-1 significantly promotes differentiation of NSCs into neurons [Citation37].
As early as the 1990s, studies using spider toxins in the treatment of central nervous system disorders have been reported [Citation23, Citation24]. In recent decades, there have been several reports on the important effects of toxins isolated and purified from spider glands on the central nervous system and the peripheral nervous system [Citation38–41]. Therefore, evaluating the effect of different concentrations of crude venom on the proliferation and differentiation of NSCs cultured in vitro and assessing the possibility of finding active factors that contribute to the proliferation and differentiation of NSCs from crude venoms of spiders is the purpose and significance of our study. From the results of the study, we can confirm that in C. jingzhao crude venom, there are components that are conducive to the proliferation and differentiation of NSCs. Determining the precise mechanism of the observed effects on the differentiation and proliferation exerted by venom treatment and separation, and identification of the accurate factors that contribute to cell differentiation and proliferation will be the directions and priorities for our next study.
Conclusions
In summary, the present study investigated the effects of varying concentrations of C. jingzhao venom on the differentiation and proliferation of C17.2 NSCs. Cells were exposed to three different venom concentrations (1, 10 and 20 μg/mL) under either differentiation or proliferation conditions. Low concentrations (1, 10 and 20 μg/mL) of crude drug had no obvious inhibitory effect on cell proliferation. In contrast, the addition of crude poison promoted the differentiation of NSC into neuronal cells. C17.2 cells cultured in differentiation medium with a venom concentration of 10 μg/mL exhibited enhanced neuronal differentiation. Addition of C. jingzhao crude venom promoted the differentiation of NSCs into neurons to a certain extent but did not inhibit the proliferation of NSCs. This result suggests that it is possible to screen one or more NGF-like polypeptides using the C. jingzhao crude venom. NGF-like polypeptides have the dual biological functions of providing neuronal nutrition and promoting the protrusive growth necessary for cultivation of NSCs. Combined with cell transplantation technology, these methods may prove feasible for treatment of senile dementia, neurasthenia and spinal cord injury.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Coddington JA, Levi HW. Systematics and evolution of spiders (Araneae). Ann Rev Ecol Syst. 1991;22:565–592.
- Shear WA, Palmer JM, Coddington JA, et al. A Devonian spinneret: early evidence of spiders and silk use. Science. 1989;246:479–481.
- King GF. The wonderful world of spiders: preface to the special Toxicon issue on spider venoms. Toxicon. 2004;43:471–475.
- Saez NJ, Senff S, Jensen JE, et al. Spider-venom peptides as therapeutics. Toxins. 2010;2:2851–2871.
- Parks TN, Mueller AL, Artman LD, et al. Arylamine toxins from funnel-web spider (Agelenopsis aperta) venom antagonize N-methyl-D-aspartate receptor function in mammalian brain. J Biol Chem. 1991;266:21523–21529.
- Donevan SD, Rogawski MA. Multiple actions of arylalkylamine arthropod toxins on the N-methyl-D-aspartate receptor. Neuroscience. 1996;70:361–375.
- McCormick KD, Kobayashi K, Goldin SM, et al. Characterization and synthesis of a new calcium antagonist from the venom of a fishing spider. Tetrahedron. 1993;49:11155–11168.
- Pereira LS, Silva PI, Miranda MTM, et al. Structural and biological characterization of one antibacterial acylpolyamine isolated from the hemocytes of the spider Acanthocurria gomesiana. Biochem Biophys Res Commun. 2007;352:953–959.
- Ashton AC, Volynski KE, Lelianova VG, et al. alpha-Latrotoxin, acting via two Ca2+-dependent pathways, triggers exocytosis of two pools of synaptic vesicles . J Biol Chem. 2001;276:44695–44703.
- Escoubas P, Bosmans F. Spider peptide toxins as leads for drug development. Expert Opin Drug Discov. 2007;2:823–835.
- Xiao Y, Tang J, Hu W, et al. Jingzhaotoxin-I, a novel spider neurotoxin preferentially inhibiting cardiac sodium channel inactivation. J Biol Chem. 2005;280:12069–12076.
- Zeng X, Deng M, Lin Y, et al. Isolation and characterization of Jingzhaotoxin-V, a novel neurotoxin from the venom of the spider Chilobrachys jingzhao. Toxicon. 2007;49:388–399.
- Mei H. Effects of the venom of the spider (Chilobrachys jingzhao sp.nov.) on the nerve, muscle and neuromuscular junction. Huazhong Normal Univ J Postgraduates. 2005;1:131–134.
- Piccini P, Brooks DJ, Björklund A, et al. Dopamine release from nigral transplants visualized in vivo in a Parkinson's patient. Nat Neurosci. 1999;2:1137–1140.
- Studer L, Tabar V, McKay R. Transplantation of expanded mesencephalic precursors leads to recovery in parkinsonian rats. Nat Neurosci. 1998;1:290–295.
- Freed CR, Greene PE, Breeze RE, et al. Transplantation of embryonic dopamine neurons for severe Parkinson's disease. N Engl J Med. 2001;344:710–719.
- Jones KR, Reichardt LF. Molecular cloning of a human gene that is a member of the nerve growth factor family. Proc Natl Acad Sci USA. 1990;87:8060–8064.
- Maisonpierre PC, Le Beau MM, Espinosa R, et al. Human and rat brain-derived neurotrophic factor and neurotrophin-3: gene structures, distributions, and chromosomal localizations. Genomics. 1991;10:558–568.
- Lee R, Kermani P, Teng KK, et al. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945–1948.
- Doetsch F, Petreanu L, Caille I, et al. EGF converts transit-amplifying neurogenic precursors in the adult brain into multipotent stem cells. Neuron. 2002;36:1021–1034.
- Chen B-Y, Wang X, Wang Z-Y, et al. Brain-derived neurotrophic factor stimulates proliferation and differentiation of neural stem cells, possibly by triggering the Wnt/β-catenin signaling pathway. J Neurosci Res. 2013;91:30–41.
- Xuan AG, Long DH, Gu HG, et al. BDNF improves the effects of neural stem cells on the rat model of Alzheimer's disease with unilateral lesion of fimbria-fornix. Neurosci Lett. 2008;440:331–335.
- Hanin I. Cholinergic toxins and Alzheimer's disease. Ann N Y Acad Sci. 1992;648:63–70.
- Saccomano NA (Ledyard, CT), Volkmann RA. (Mystic, CT), inventor; Pfizer Inc. (New York, NY), assignee. Indolyl-3 polyamines and their use as antagonists of excitatory amino acid neurotransmitters. United States patent 5037846; 1991.
- Snyder EY, Deitcher DL, Walsh C, et al. Multipotent neural cell lines can engraft and participate in development of mouse cerebellum. Cell.1992;68:33–51.
- Lundqvist J, El Andaloussi-Lilja J, Svensson C, et al. Optimisation of culture conditions for differentiation of C17.2 neural stem cells to be used for in vitro toxicity tests. Toxicol In Vitro. 2013;27:1565–1569.
- Attoff K, Kertika D, Lundqvist J, et al. Acrylamide affects proliferation and differentiation of the neural progenitor cell line C17.2 and the neuroblastoma cell line SH-SY5Y. Toxicol In Vitro. 2016;35:100–111.
- Wang G, Ao Q, Gong K, et al. The effect of topology of chitosan biomaterials on the differentiation and proliferation of neural stem cells. Acta Biomater. 2010;6:3630–3639.
- Huang H, Liu L, Li B, et al. Ketamine interferes with the proliferation and differentiation of neural stem cells in the subventricular zone of neonatal rats. Cell Physiol Biochem. 2015;35:315–325.
- Han SSW, Kang DY, Mujtaba T, et al. Grafted lineage-restricted precursors differentiate exclusively into neurons in the adult spinal cord. Exp Neurol. 2002;177:360–375.
- Cattaneo E, McKay R. Proliferation and differentiation of neuronal stem cells regulated by nerve growth factor. Nature. 1990;347:762–765.
- Huang L, Wang G. The effects of different factors on the behavior of neural stem cells. Stem Cell Int. 2017;9497325 [16 p]. DOI:10.1155/2017/9497325
- Johe KK, Hazel TG, Muller T, et al. Single factors direct the differentiation of stem cells from the fetal and adult central nervous system. Gen Dev. 1996;10:3129–3140.
- Cameron HA, Hazel TG, McKay RDG. Regulation of neurogenesis by growth factors and neurotransmitters. J Neurobiol. 1998;36:287–306.
- Arsenijevic Y, Weiss S, Schneider B, et al. Insulin-like growth factor-1 is necessary for neural stem cell proliferation and demonstrates distinct actions of epidermal growth factor and fibroblast growth factor-2. J Neurosci. 2001;21:7194–7202.
- Yan YH, Li SH, Gao Z, et al. Neurotrophin-3 promotes proliferation and cholinergic neuronal differentiation of bone marrow-derived neural stem cells via notch signalling pathway. Life Sci. 2016;166:131–138.
- Lööv C, Fernqvist M, Walmsley A, et al. Neutralization of LINGO-1 during in vitro differentiation of neural stem cells results in proliferation of immature neurons. PLoS One. 2012:7:e29771.
- Cristofori-Armstrong B, Rash LD. Acid-sensing ion channel (ASIC) structure and function: Insights from spider, snake and sea anemone venoms. Neuropharmacology. 2017;127:173–184.
- Joeres N, Augustinowski K, Neuhof A, et al. Functional and pharmacological characterization of two different ASIC1a/2a heteromers reveals their sensitivity to the spider toxin PcTx1.Sci Rep UK. 2016:27647 [14 p].
- McCarthy CA, Rash LD, Chassagnon IR, et al. PcTx1 affords neuroprotection in a conscious model of stroke in hypertensive rats via selective inhibition of ASIC1a. Neuropharmacology. 2015;99:650–657.
- Osmakov DI, Koshelev SG, Andreev YA, et al. Conversed mutagenesis of an inactive peptide to ASIC3 inhibitor for active sites determination. Toxicon. 2016;116:11–16.