
Abstract
Neolamarckia cadamba is a miracle tree species with considerable economic potential used as a timber wood and traditional medicine resource in South and Southeast Asia. To better understand the molecular basis of its chloroplast biology, we sequenced and characterised the complete chloroplast genome using Illumina pair-end sequencing. The analysis showed a chloroplast genome size of 154,999 bp in length, harbouring a pair of inverted repeats (IRs) of 25,634 bp separated by a large single copy (LSC) sequence of 85,880 bp and a small single copy (SSC) sequence of 17,851 bp. Of the 130 genes present, 96 were unique and 17 were duplicated in the IRs. The coding regions comprised 79 protein genes, 30 tRNA genes and 4 rRNA genes. In the protein-coding genes, nine genes contained one intron each, while another two genes comprised two introns. The overall GC content of the chloroplast genome was 37.6%. Simple sequence repeat (SSR) analysis revealed that most SSRs were A/T-rich, which contributed to the overall AT richness of the N. cadamba chloroplast genome. Additionally, fewer SSRs distributed in the protein-coding sequences compared to the non-coding regions, indicating an uneven distribution of SSRs within the chloroplast genome. A maximum likelihood phylogenetic analysis showed that N. cadamba was closely related to Morinda officinalis and Emmenopterys henryi, which belong to the Rubiaceae family. This complete chloroplast genome will offer valuable information for the development of highly variable DNA markers for future conservation, genetic engineering studies and variety selection of this miracle tree.
Introduction
Neolamarckia cadamba is a tropical deciduous tree that grows mainly in South and Southeast Asia [Citation1]. As a fast-growing tree with anatomical, morphological, and chemical characteristics, N. cadamba has tremendous economic and ecological value [Citation2]. For example, the bark, leaf and flower of N. cadamba are widely used in the treatment of several diseases, including but not limited to fever, diarrhoea and elephantiasis [Citation3]. More than 50 compounds have been isolated and structurally identified from N. cadamba to date. Phytochemistry analysis released that the stem bark of N. cadamba contains tannins, saponins, steroids, alkaloids, quinovic acid, cadambagenic acid and β-sitosterol [Citation4,Citation5]. The major phytochemicals present in the leaves of N. cadamba are alkaloids, flavonoids, steroids and glycosides [Citation6]. The essential oil from flowers contains gerenyl acetate, geraniol, linalool, linalyl acetate, α-selinene, 2-nonanol and β-phellandrene as major compounds [Citation7]. The heartwood contains cadambine [Citation8]. Modern pharmacological research has demonstrated that most of these compounds have multiple important and desirable therapeutic actions, including antipyretic, antioxidant, antimicrobial and antilipidemic activities [Citation9]. With this evidence, it is no wonder that N. cadamba has been an ayurvedic remedy in many Indian traditional medicines [Citation10]. In south China, N. cadamba is currently being cultivated for urban greening, building materials, pulp production and animal feed ingredients [Citation11]. Hence, it is also known as the ‘miracle tree’. In addition to the significant medicinal and ecological value describe above, N. cadamba is exemplary for its relatively small genome size (∼800 Mb), fast growth, blossoming and yielding fruits within four years. These characteristics make N. cadamba an exemplary starting point to investigate the mechanism of its secondary metabolism.
In the genomic and post-genomic era, the mining of plant chloroplast genome is now of interest. Interestingly, the chloroplast genome usually has a typical quadripartite structure with a pair of inverted repeats (IRs) separated by a large single-copy (LSC) region and a small single-copy (SSC) region. The chloroplast genome ranges from 120 to 160 Kb in length and exhibits highly conserved gene order and contents [Citation12,Citation13]. In angiosperms, the chloroplast genome contains approximately 130 genes which encode an array of proteins in relation to photosynthesis, nitrogen fixation and biosynthesis of starch, pigments, fatty acids and amino acids [Citation14,Citation15]. The most common and important protein is Rubisco, which is probably also the most abundant protein in plant cells. Rubisco is an enzyme involved in carbon fixation in the photosynthesis process. It is worth mentioning that in addition to Rubisco, the chloroplast genome encodes low molecular mass subunits of Photosystem II which contain light-harvesting complexes with chlorophyll and carotenoids that absorb light energy and use it to energize electrons [Citation14]. In contrast to nuclear genome, chloroplast genome exhibits a much smaller size for complete sequencing and shows high copy numbers in each cell, conferring the ability to be an attractive site for high level expression of transgenes. Recent efforts have demonstrated the potential to address phylogenetic questions at or above the species level, and understand structural and functional evolution by using the whole chloroplast genome sequences [Citation16–18]. In the genetic engineering field, chloroplast transformation in higher plants has been shown to have a variety of advantages such as the absence of gene silencing, site-specific integration of transgenes via homologous recombination within the chloroplast genome, and strictly maternal inheritance that avoids dissemination of transgenes by pollen [Citation19, Citation20]. These characteristics make chloroplast transformation a powerful tool for the creation of genetically modified crops with expected improved traits.
To date, few data are available regarding the N. cadamba genome. Here, as a part of the genome sequencing project of N. cadamba, we report its complete chloroplast genome sequence, determined using the Illumina sequencing platform. Information on the complete chloroplast genome will be instrumental for genetic engineering and breeding programmes. The comparative sequence analysis will also contribute to a better understanding of the evolution within the Rubiaceae family.
Materials and methods
DNA sequencing, genome assembly and validation
Fresh young leaves from one individual of N. cadamba were collected from South China Agricultural University, Guangdong province of China. High quality DNA was obtained as follows: chloroplast was isolated from 5 g fresh leaves using the method introduced by Hao et al. [Citation21] with a few modifications. All steps were conducted at 4 °C unless otherwise specified. The chloroplast pellets were lysed by adding 2.0 mL extraction buffer and incubated at 50 °C for 2 h. The crude cytoplasmic DNA was washed twice with 70% ethanol and resuspended in ddH2O buffer with RNase digestion for 30 min at 37 °C. From the DNA sample, a 500-bp paired-end library was constructed, and approximately 2 Gb of raw sequence with an average read length of 300 bp was generated on the Illumina Hiseq X ten PE150 platform. Prior to de novo genome assembly, raw reads were mapped to the NCBI Viridiplantae chloroplast genomes using BWA to filter the non-chloroplastic reads [Citation22] in order to avoid contamination of mtDNA and nuclear DNA. Subsequently the filtered reads were assembled into non-redundant contigs with SOAP de novo (version 2.04). All contigs were then mapped to the reference chloroplast genome of Coffea arabica L. (GenBank No. EF044213) [Citation23]. Finally, additional PCR amplifications and Sanger sequencing were used to fill the gaps between the contigs, as well as to verify the junctions between the LSC/SSC and the IRs regions. The primer for LSC-IRa was (LSC/IRa-F: TTT GTG ACC TAC CAT ACG AT, and LSC/IRa-R: ACC CTG TAG ACC ATC CTC AT), for IRa-SSC it was (IRa/SSC-F: CCA CTT TCA CTT TAC AGA GAC AG, and IRa/SSC-R: AAT GGG GTT GGT GTT GTT AG), for SSC-IRb (SSC/IRb-F: CCA TCA CTT TCG CCT ATC TC, and SSC/IRb-R: CAC TTT CAC TTT ACA GAG ACA GG) and for IRb-LSC (IRb/LSC-F: TCA GCA ACA GTC GGA CAA GT, and IRb/LSC-R: TTA GAG TAG AGG GCG GAT GTA G).
Genome annotation and sequence statistics
The chloroplast genome was annotated using the online program CpGAVAS coupled with manual correction for protein-coding regions boundaries [Citation24]. The tRNA genes were identified using tRNAscan-SE 1.21 [Citation25]. The nomenclature of chloroplast genes was referred to the Chloroplast DB. The circular chloroplast genome map was drawn using the OGDRAW program [Citation26]. GC content and codon usage were analysed using MEGA5.
Genome comparison and repeat content
The complete chloroplast genome of N. cadamba was compared with the chloroplast genomes of C. arabica (GenBank No. EF044213), Coffea canephora (GenBank No. KU500324), Emmenopterys henryi (GenBank No. KY273445) and Morinda officinalis (GenBank No. KR869730) using mVISTA in Shuffle-LAGAN mode and Blast Ring Image Generator with default parameters [Citation27]. M. officinalis was set as a reference. The comparison of IR/LSC and IR/SSC regions was conducted using the GenBank genome files of C. arabica, C. canephora, E. henryi and M. officinalis with coordinates for the gene features. For the identification of perfect and compound simple sequence repeats (SSRs), the Perl script MISA was used with a motif size of one to six nucleotides and thresholds of eight, four, four, three, three, and three repeat units for mono-, di-, tri-, tetra-, penta-, and hexa nucleotide stretches as SSRs. For the identification of the dispersed repeats including the forward, reverse and palindromic repeats, REPUTER was used with a minimal size of 30 bp and >90% identity (Hamming distance equal to 3) between the two repeat copies, and the identified repeat regions were checked with the corresponding genomic coordinates and were annotated [Citation28]. All of the repeats identified with the various programs were manually verified to remove redundant results.
Sequence divergence and phylogenetic analysis
To elucidate the N. cadamba phylogenetic position within the Rubiaceae family, multiple alignments were performed using 79 protein-coding genes shared by the chloroplast genomes of five Rubiaceae members representing four genera. In addition, 15 species from the neighbouring families (i.e. Gentianaceae and Apocynaceae) were downloaded from NCBI Organelle Genome Resources database. The 79 protein-coding gene sequences were aligned using the Clustal algorithm. Pairwise sequence divergences were calculated using Kimura’s two-parameter (K2P) model. Maximum likelihood (ML) analysis was performed using RAxML 8.0 software with the GTR + I + G nucleotide substitution model [Citation29]. This appropriate model of DNA sequence evolution was determined by Modeltest 3.7. Furthermore, Mrmodeltest2.3 was used to determine the best-evolution for the data set for Bayesian inference [Citation30]. Bayesian inference was calculated using MrBayes3.1.2 with a general time reversible model of DNA substitution and a gamma distribution rate variation across sites [Citation31]. Branches that received bootstrap support for ML and Bayesian posterior probabilities (BPP) ≥ 75% (ML) and 0.95 (BPP), respectively, were considered as significantly supported.
Results and discussion
Chloroplast genome organisation and features of N. cadamba
The complete chloroplast genome of N. cadamba (submitted to GenBank under accession MG572117) was 154,999 bp in length and had the common quadripartite structure found in most land plants. The genome included an LSC of 85,880 bp (covering 55.4%) and a SSC of 17,851 bp (covering 11.5%), separated by two IRs of 25,634 bp (covering 33.1%) (). The overall GC content of the N. cadamba chloroplast genome was 37.6%, with a higher GC content in the IRs regions (43.2%) than in LSC and SSC (35.4 and 31.6%, respectively). This high GC percentage in the IR regions was due to the presence of eight ribosomal RNA (rRNA) sequences in these regions. These results were in agreement with the previously reported high GC percentage in the IR regions, which could be attributed to the presence of ribosomal RNA [Citation32]. In addition, the GC content of the N. cadamba chloroplast genome was higher than that of C. arabica (37.00%); similar to that of C. canephora (37.47%) and E. henryi (37.64%); and lower than that of M. officinalis (38.05%) (). Most of the chloroplast genome sequence encoded proteins, tRNAs and rRNAs (51.64, 1.98 and 6.02%, respectively). The remaining regions were non-CDSs, including introns, intergenic spacers and pseudogenes. A total of 130 genes were identified in the N. cadamba chloroplast genome, including 79 protein-coding genes, 30 tRNAs genes, 4 rRNAs, 2 pseudogenes ( and ). Of the observed gene space in N. cadamba, 73 protein-coding genes and 23 tRNA were found to be unique, whereas six protein-coding (rpl2, rpl23, ycf2, ndhB, rps7 and rps12), seven tRNAs (trnI-CAU, trnL-CAA, trnV-GAC, trnI-GAU, trnA-UGC, trnR-ACG and trnN-GUU) and four rRNA genes (rrn4.5, rrn5, rrn16 and rrn23) were found be duplicated in IRa and IRb (). The gene rps12 was trans-spliced and its 5′ end exon located in the LSC region, while the 3′ exon and intron were duplicated and located in the IR regions. Twelve protein-coding genes and six tRNAs had introns, 15 with one intron and three with two introns. A recent study has demonstrated that the presence of introns could improve exogenous gene expression at specific times and positions, resulting in the expected agronomic traits. Therefore, these introns can be the potential sites to improve the transformation efficiency [Citation33]. In addition, a portion of the rps19 gene was duplicated at the IRa–LSC boundary as a result of expansion of the IR. A similar pattern of this gene was also observed in C. arabica [Citation23].
Figure 1. Chloroplast genome of N. cadamba. Note: Genes inside the circle are transcribed clockwise; genes outside are transcribed counter-clockwise. The dark grey inner circle corresponds to the GC content; the light-grey to the AT content.
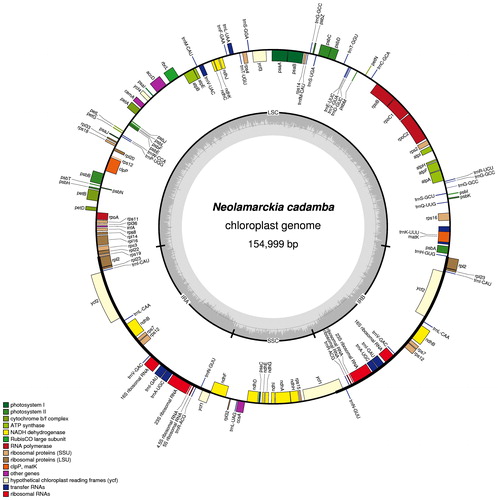
Table 1. Summary of complete chloroplast genomes for five Rubiaceae species.
Table 2. List of genes encoded by N. cadamba chloroplast genome.
Table 3. SSRs distribution in the five Rubiaceae chloroplast genomes.
Comparative analysis of five chloroplast genomes of Rubiaceae species
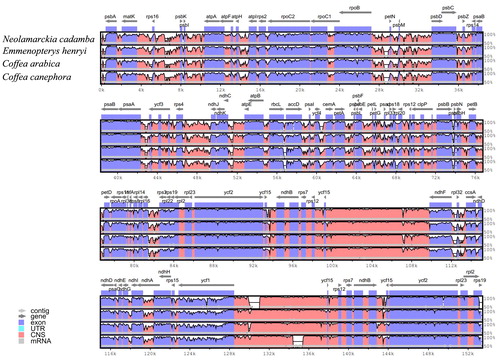
In this study, four complete chloroplast genomes within the Rubiaceae family, namely C. arabica [Citation23], C. canephora [Citation34], E. henryi [Citation35], and M. officinalis [Citation36] were selected for comparison with N. cadamba. The genome size of E. henryi was the largest among these plants, and this difference was mostly attributed to the variation in the length of the SSC region (). Pairwise chloroplast genomic alignment between N. cadamba and four other species revealed a high degree of synteny, suggesting the evolutionary conservation of these genomes at the genome-scale level. Furthermore, a multiple sequence alignment based on mVISTA was performed among these species chloroplast genomes for investigating the levels of sequence divergence between them. The results showed that the whole chloroplast genomes were rather conservative, although some divergent regions were found as well. For example, the LSC and SSC regions were more divergent than the two IR regions. Furthermore, the non-coding regions exhibited higher divergence than the coding regions (). The most dissimilar coding regions include ndhH, ndhF, matK, ycf1, rbcL, ccsA and accD. Similar results have also been observed in other flowering plants [Citation37,Citation38]. Therefore, these divergence hotspot regions may facilitate the development of molecular markers for phylogenetic and phylogeographic analyses as well as the species identification of Rubiaceae representatives.
Figure 2. Sequence identity plots (mVISTA) among five species of Rubiaceae, using Morinda officinalis as reference genome. Note: Grey arrows above the alignment indicate genes with their orientation. A cut-off of 70% identity was used for the plots, and the Y-scale represents the percent identity between 50% and 100%.
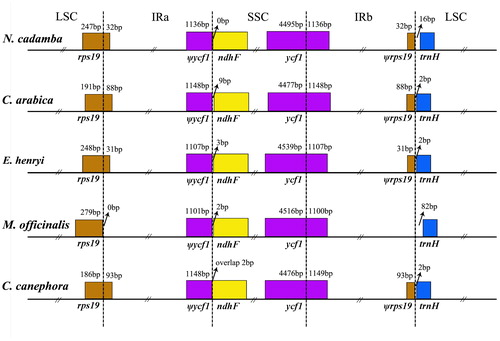
Although IRs are highly conserved among land plants and accumulate few mutations, the expansion and contraction at the borders of the IR regions often results in size variation of chloroplast genomes [Citation39]. The IRs border positions and their adjacent genes in N. cadamba chloroplast genome were compared to the corresponding regions among these species (). Although the overall genomic structure, including gene number and gene order, was well-conserved, the five Rubiaceae chloroplast genomes exhibited obvious differences at the IR/SC boundary regions (). The IR region expanded into the rps19 gene, creating a pseudogene fragment ψrps19 at the IRa/LSC border with lengths of 31–93 bp in four species (i.e. C. arabica, C. canephora, E. henryi and N. cadamba). In all species, the IRb/SSC borders extended into the ycf1 genes to create long ycf1 pseudogenes with variable length, as observed in other species as well [Citation40]. The length of the ycf1 pseudogene is 1136 bp in N. cadamba, 1148 bp in C. arabica, 1148 bp in C. canephora, 1107 bp in E. henryi and 1101 bp in M. officinalis. In addition, the ndhF gene and the ψycf1 fragment overlapped by 2 bp at the junction of the IRa and SSC regions in C. canephora. However, for the other four species, the ndhF genes are all located in the SSC region (). In addition, the trnH genes were located in the LSC region, 2–82 bp apart from the IRb/LSC border.
Figure 3. Comparison of the borders of LSC, SSC and IR regions among five chloroplast genomes.
Repeat sequence analysis of N. cadamba chloroplast genome
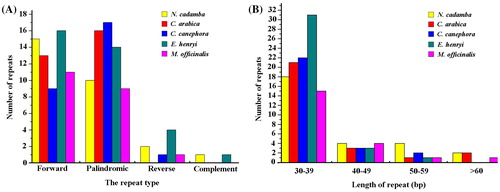
Repeat sequences play an essential role in the evolutionary processes involving mutations and genome rearrangements. They are not only considered as a hotspot for the analysis of regulatory sites in gene expression, but are also helpful in phylogenetic analyses [Citation41]. For repeat analysis, a total of 28 repeats were found in the N. cadamba chloroplast genome, including 15 forward repeats, 10 palindromic repeats, two reverse repeats and one complement repeat (). These repeats were found to be at least 30 bp per unit and the biggest was 75 bp. Among these, eight forward repeats had a size of 30–39 bp in length, whereas seven palindromic repeats were found to be same length, two reverse repeats and one complement repeat were 31 bp in length (). Besides, two palindromic repeats and two forward repeats were 40–49 bp in length, and the rest were >50 bp (). Similarly, 29, 27, 35, 21 repeat pairs were found in previously reported C. arabica, C. canephora, E. henryi and M. officinalis genomes (), respectively, when compared with N. cadamba (). About one-third of these repeats were distributed in protein-coding sequences. Previous report suggested that sequence variation and genome rearrangement occur due to slipped strand mispairing and improper recombination of these repeat sequences [Citation42]. These repeats can provide important clues towards understanding the spatial-temporal organisation in Rubiaceae.
Figure 4. Analysis of repeated sequences in the five chloroplast genomes of Rubiaceae family. Total number (A) of four repeat types. Frequency (B) of the total repeats by length.
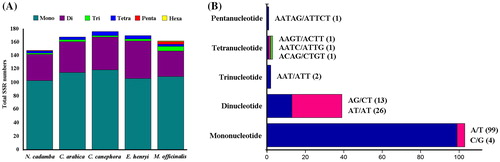
In addition to the long repeat sequences, we further explored the role of SSR in the chloroplast genome of these five Rubiaceae species. SSRs are short base-pair motifs which are repeated many times in tandem. These sequences are commonly distributed in untranslated regions throughout the genome. The frequent mutations in SSR regions are easily to alter the number of repeats, result in a higher degree of polymorphism. Because of these characteristics, SSRs have become important molecular markers for a wide range of applications [Citation43,Citation44]. In compliance with other similar studies [Citation32,Citation45], we detected the perfect SSRs longer than 8 bp because SSRs of 8 bp or longer are prone to slip-strand mispairing, which is thought to be the primary mutational mechanism causing their high level of polymorphism. In our analysis, the total number of SSRs ranged from 148 in N. cadamba to 176 in C. canephora (). The majority of SSRs in all species were mononucleotides, varying in quantity from 103 in N. cadamba to 119 in C. canephora. Dinucleotides were the second most prevalent type, ranging in quantity from 38 in M. officinalis to 56 in E. henryi. Generally, the trinucleotide, tetranucleotide, pentanucleotide and hexanucleotide repeat units are rare in the chloroplast genome, and only two trinucleotides, three tetranucleotides and one pentanucleotides repeat units were observed in the N. cadamba chloroplast genome. It is noteworthy that the majority of tri- to hexanucleotides were AT-rich in all species. Moreover, an average of 67.15% (69.59% in N. cadamba) of the SSRs were A/T mononucleotides in these chloroplast genomes, slightly lower than the 76% previously reported for monocot chloroplast genomes [Citation32]. Our finding is in agreements with previous reports that chloroplast SSRs generally contain polyadenine or polythymine repeats [Citation46]. Thus, these SSRs contribute to the AT richness of the Rubiaceae chloroplast genomes, as previously reported for different species [Citation32]. SSRs were also detected in the CDS regions of the N. cadamba chloroplast genome. Interestingly, although protein-coding genes account for approximately 50% of the total length of the chloroplast genome, only 35% of the SSRs exist within these regions. This suggests that SSRs were less abundant in CDS than in non-coding regions and that they are unevenly distributed within the chloroplast genomes. In total, 61 SSRs were identified in the CDS of 25 genes in N. cadamba. Among them, 13 genes were found to contain at least two SSRs, including atpF, matK, ndhD, rpoC2, ycf1, ycf2, etc. These identified SSRs could be used for determination of the phylogeography and population structure pattern of N. cadamba in future studies.
Figure 5. Analysis of simple sequence repeat in the five chloroplast genomes of Rubiaceae family. Number (A) of different SSRs types detected in five genomes. Frequency (B) of different SSR types in N. cadamba.
Potential sites for chloroplast genetic engineering
The chloroplast is an ideal host for expression of transgenes for its unique advantages including the transgene stacking, high levels of transgene expression, lack of gene silencing and large amounts of foreign protein accumulation [Citation47]. Significant progress in chloroplast transformation has been made in the model species tobacco as well as in a few major crops [Citation48,Citation49]. Usually, the intergenic spacer regions of chloroplast genome are used as gene introduction sites for vector construction. However, the transformation efficiency is affected when the sequences for homologous recombination are divergent among different species. In order to develop a universal plastid transformation vector for Rubiaceae family members, the most commonly used sites including trnI/trnA and accD/rbcL were compared among five species. The results show that the trnI/trnA regions exhibit higher level of homology among these plant species, even the exons of these genes are complete homologous. Therefore, this optimal intergenic spacer can be selected for transgene integration and to develop site-specific chloroplast transformation vector for Rubiaceae family. In the near future, the genes related to bioactive compound synthesis will be the primary targets for investigation. In addition, using chloroplast genetic engineering to introduce useful traits, such as insect resistance and cold tolerance, might be other applications to improve these economical plants.
Phylogenetic analysis
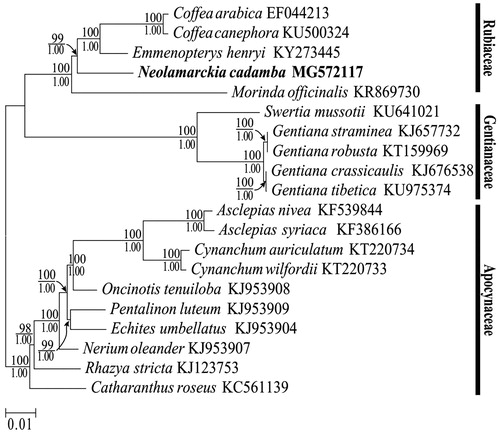
Some of the problems of phylogeny in angiosperms and closely related species have found solutions owing to the advancements in next-generation sequencing of whole chloroplast genomes [Citation50–52]. To study the phylogenetic position of N. cadamba within the Rubiaceae family, we used 79 protein-coding genes shared by the chloroplast genomes of five Rubiaceae members, and 15 species from the neighbouring families to construct a phylogenetic tree. Multiple alignments showed extremely conserved chloroplast genome sequences within the Rubiaceae family. ML analysis revealed all of the nodes with bootstrap values ≥98%, and most of these nodes had 100% bootstrap values. Bayesian inference yielded nearly identical tree topologies across all analyses, with 1.0 BPP at each node (). Both the ML and Bayesian inference phylogenetic results strongly supported the position of N. cadamba clustered within the Rubiaceae family (). Neolamarckia was sister to Emmenopterys + Coffea, and the two genera contributed to a diverging lineage of the basal genera Morinda. Finally, the phylogenetic analyses support the placement of Rubiaceae sister to the Gentianaceae. The position of the Rubiaceae and Gentianaceae is not controversial as recent single and multigene phylogenies already strongly support their placement in the Euasterid I clade [Citation23]. Our study provides new data for deeper analysis of phylogeny within Rubiaceae family. However, existing chloroplast genome sequences of Rubiaceae remain sporadic and we need more chloroplast genome data to determine the position of different evolutionary branches. We believe that more chloroplast genomes of Rubiaceae species will be completely sequenced with the sequencing cost decreasing.
Figure 6. Maximum likelihood phylogenomic tree inferred from 20 complete chloroplast genomes. Note: Maximum likelihood bootstrap proportions (above the line) higher than 50% and Bayesian posterior probabilities (under the line) more than 0.95 were indicated along branches.
Conclusion
In summary, this study reported the complete chloroplast genome of N. cadamba (154,999 bp). The genome sequencing, assembly, annotation, and the comparative analysis, revealed that the chloroplast genomes of N. cadamba share a quadruple structure, gene order, GC content, and codon usage features, similar to previously reported chloroplast genomes from Rubiaceae family. The location and distribution of repeat sequences were detected. These results help us to explore new molecular markers and study the phylogenetic relationships of plant species. Moreover, we compared and evaluated the potential site for the design of plastid transformation vectors for this economically important family. Furthermore, ML and BI phylogenetic trees were constructed on the basis of protein-coding genes, which were also shared by four Rubiaceae members from three genera. This phylogenomic analysis provided the first successful attempt to clarify intrageneric relationships within Rubiaceae.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Huang H, Li JC, Ouyang KX, et al. Direct adventitious shoot organogenesis and plant regeneration from cotyledon explants in Neolamarckia cadamba. Plant Biotech. 2014;31(2):115–121.
- Lal M, Dutt D, Tyagi CH, et al. Characterization of Anthocephalus cadamba and its delignification by kraft pulping. Tappi J. 2010;9(3):30–37.
- Pandey A, Negi PS. Traditional uses, phytochemistry and pharmacological properties of Neolamarckia cadamba: a review. J Ethnopharmacol. 2016;181:118–135.
- The Ayurvedic Pharmacopoeia of India. New Delhi, India: Department of AYUSH, Ministry of Health and Family Welfare, Govt. of India, Part I, vol. II, 1st ed.; 2011.
- Chandel M, Sharma U, Kumar N, et al. In vitro studies on the antioxidant/antigenotoxic potential of aqueous fraction from Anthocephalus cadamba bark. In: Sudhakaran PR, editor. Perspectives in cancer prevention—translational cancer research. New Delhi, India: Springer; 2014. p. 61–72.
- Kumar A, Chowdhury SR, Jatte KK, et al. Anthocephaline, a new indole alkaloid and cadambine, a potent inhibitor of DNA topoisomerase IB of Leishmania donovani (LdTOP1LS), isolated from Anthocephalus cadamba. Nat Prod Commun. 2015;10(2):297–299.
- Khare CP. Indian herbal remedies: rational western therapy, ayurvedic and other traditional usage, botany. New York, USA: Springer; 2011. p. 66–67.
- Jeyalalitha T, Murugan K, Umayavalli M. Preliminary phytochemical screening of leaf extracts of Anthocephalus cadamba. Int J Recent Sci Res. 2015;6:6608–6611.
- Pandey A, Negi PS. Phytochemical composition, in vitro antioxidant activity and antibacterial mechanisms of Neolamarckia cadamba fruits extracts. Nat Prod Res. 2017;32(10):1189–1192.
- Nanda GC, Dash SK, Das I. Screening of vishaghna (antitoxic) plants in Ayurveda. Indian J Pharm Sci Res. 2014;4:43–48.
- Li JC, Hu XS, Huang XL, et al. Functional identification of an EXPA gene (NcEXPA8) isolated from the tree Neolamarckia cadamba. Biotechnol Biotechnol Equip. 2017;31(6):1116–1125.
- Sugiura M. The chloroplast genome. Plant Mol Biol. 1992;19(1):149–168.
- Wicke S, Schneeweiss GM, Müller KF, et al. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol. 2011;76(3–5):273–297.
- Neuhaus HE, Emes MJ. Nonphotosynthetic metabolism in plastids. Annu Rev Plant Physiol Plant Mol Biol. 2000;51(51):111–140.
- Liu J, Qi ZC, Zhao YP, et al. Complete cpDNA genome sequence of Smilax china and phylogenetic placement of Liliales-influences of gene partitions and taxon sampling. Mol Phylogenet Evol. 2012;64(3):545–562.
- Zhang YX, Iaffaldano BJ, Zhuang XF, et al. Chloroplast genome resources and molecular markers differentiate rubber dandelion species from weedy relatives. BMC Plant Biol. 2017;17(1):34. DOI:10.1186/s12870-016-0967-1.
- Asaf S, Waqas M, Khan AL, et al. The complete chloroplast genome of wild rice (Oryza minuta) and its comparison to related species. Front Plant Sci. 2017;8:304. DOI:10.3389/fpls.2016.00843.
- Kaila T, Chaduvla PK, Rawal HC, et al. Chloroplast genome sequence of clusterbean (Cyamopsis tetragonoloba L.): genome structure and comparative analysis. Genes. 2017;8(9):e212. DOI:10.3390/genes8090212.
- Zienkiewicz M, Krupnik T, Drożak A, et al. Transformation of the Cyanidioschyzon merolae chloroplast genome: prospects for understanding chloroplast function in extreme environments. Plant Mol Biol. 2017;93(1–2):171–183.
- Li D, Han X, Zuo J, et al. Construction of rice site-specific chloroplast transformation vector and transient expression of EGFP gene in Dunaliella salina. J Biomed Nanotechnol. 2011;7(6):801. DOI:10.1166/jbn.2011.1339.
- Hao W, Fan S, Hua W, et al. Effective extraction and assembly methods for simultaneously obtaining plastid and mitochondrial genomes. PLoS ONE. 2014;9(9):e108291. DOI:10.1371/journal.pone.0108291.
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595.
- Samson N, Bausher MG, Lee SB, et al. The complete nucleotide sequence of the coffee (Cofea arabica L.) chloroplast genome: organization and implications for biotechnology and phylogenetic relationships amongst angiosperms. Plant Biotechnol J. 2007;5(2):339–353.
- Liu C, Shi LC, Zhu YJ, et al. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genomics. 2012;13(1):715. DOI:10.1186/1471-2164-13-715.
- Schattner P, Brooks AN, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005;33:686–689.
- Lohse M, Drechsel O, Kahlau S, et al. Organellar genome DRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013;41:W575–W581.
- Frazer KA, Pachter L, Poliakov A, et al. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004;32:W273–W279.
- Kurtz S, Choudhuri JV, Ohlebusch E, et al. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2011;29(22):4633–4642.
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313.
- Nylander JAA. MrModeltest v2. Program distributed by the author. Uppsala (Sweden): Evolutionary Biology Centre, Uppsala University; 2004.
- Ronquist F, Huelsenbeck JP. MRBAYES 3: bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574.
- Qian J, Song JY, Gao HH, et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE. 2013;8(2):e57607. DOI:10.1371/journal.pone.0057607.
- Xu JW, Feng DJ, Song GS, et al. The first intron of rice EPSP synthase enhances expression of foreign gene. Sci China C Life Sci. 2003;46(6):561–569.
- Wu DY, Bi CW, Wang XL, et al. The complete chloroplast genome sequence of an economic plant Coffea canephora. Mitochondrial DNA B. 2017;2(2):483–485.
- Duan RY, Huang MY, Yang LM, et al. Characterization of the complete chloroplast genome of Emmenopterys henryi (Gentianales: Rubiaceae), an endangered relict tree species endemic to China. Conserv Genet Resour. 2017;9(3):1–3.
- Zhang RJ, Li Q, Gao JL, et al. The complete chloroplast genome sequence of the medicinal plant Morinda officinalis (Rubiaceae), an endemic to China. Mitochondrial DNA A. 2016;27(6):4324–4325.
- Chen JH, Hao ZD, Xu HB, et al. The complete chloroplast genome sequence of the relict woody plant Metasequoia glyptostroboides Hu et Cheng. Front Plant Sci. 2015;6:447. DOI:10.3389/fpls.2015.00447.
- Dong WP, Liu J, Yu J, et al. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE. 2012;7:e35071. DOI:10.3389/fpls.2015.00447.
- Li Z, Long HX, Zhang L, et al. The complete chloroplast genome sequence of tung tree (Vernicia fordii): organization and phylogenetic relationships with other angiosperms. Sci Rep. 2017;7:1869. DOI:10.1038/s41598-017-02076-6.
- Zhou JG, Chen XL, Cui YX, et al. Molecular structure and phylogenetic analyses of complete chloroplast genomes of two Aristolochia medicinal species. Int J Mol Sci. 2017;18(9):1839. DOI:10.3390/ijms18091839.
- Asaf S, Khan AL, Khan MA, et al. Chloroplast genomes of Arabidopsis halleri ssp. gemmifera and Arabidopsis lyrata ssp. petraea: structures and comparative analysis. Sci Rep. 2017;7:7556. DOI:10.1038/s41598-017-07891-5.
- Timme RE, Kuehl JV, Boore JL, et al. A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: identification of divergent regions and categorization of shared repeats. Am J Bot. 2004;94(3):302–312.
- Thakur AK, Singh KH, Singh L, et al. SSR marker variations in Brassica species provide insight into the origin and evolution of Brassica amphidiploids. Hereditas. 2018;155(1):6. DOI:10.1186/s41065-017-0041-5.
- Kirungu JN, Deng YF, Cai XY, et al. Simple sequence repeat (SSR) genetic linkage map of D genome diploid cotton derived from an interspecific cross between Gossypium davidsonii and Gossypium klotzschianum. Int J Mol Sci. 2018;19(1):204. DOI:10.3390/ijms19010204.
- Taheri S, Lee AT, Yusop MR, et al. Mining and development of novel SSR markers using next generation sequencing (NGS) data in plants. Molecules. 2018;23(2):399. DOI:10.3390/molecules23020399.
- Kuang DY, Wu H, Wang YL, et al. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): implication for DNA barcoding and population genetics. Genome. 2011;54(8):663–673.
- Verma D, Samson NP, Koya V, et al. A protocol for expression of foreign genes in chloroplasts. Nat Protoc. 2008;3(4):739–758.
- Verma D, Daniell H. Chloroplast vector systems for biotechnology applications. Plant Physiol. 2007;145(4):1129–1143.
- Lee SM, Kang K, Chung H, et al. Plastid transformation in the monocotyledonous cereal crop, rice (Oryza sativa) and transmission of transgenes to their progeny. Mol Cells. 2006;21(3):401–410.
- Ng PK, Lin SM, Lim PE, et al. Complete chloroplast genome of Gracilaria firma (Gracilariaceae, Rhodophyta), with discussion on the use of chloroplast phylogenomics in the subclass Rhodymeniophycidae. BMC Genomics. 2017;18(1):40. DOI:10.1186/s12864-016-3453-0.
- Huang H, Shi C, Liu Y, et al. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol Biol. 2014;14(1):151. DOI:10.1186/1471-2148-14-151.
- Asaf S, Khan AL, Khan AR, et al. Complete chloroplast genome of Nicotiana otophora and its comparison with related species. Front Plant Sci. 2016;7:843. DOI:10.3389/fpls.2016.00843.