
Abstract
This study presents a seamless, efficient and restriction/ligation-independent cloning strategy for inserting large fragments: MCT cloning (based on the generation of Manmade Cohesive Termini). This method requires only four steps: (i) the first parallel PCR for the amplification of complete and truncated insert fragments; (ii) the second parallel PCR for the exponential amplification of linear plasmids with the same sequences and different break sites mediated by the first products and the corresponding reverse primers; (iii) the formation of manmade cohesive termini after DpnI digestion, mixing, denaturation and annealing; and (iv) transformation. By employing this strategy, large fragments (1–4 kbp) can be easily inserted into the pGADT7 vector (∼ 8 kbp).
Keywords:
Introduction
Molecular cloning is a fundamental and essential technique in biological research. Traditional cloning methods often rely on restriction enzymes and DNA ligase during the cloning process, which requires specific sites or sequences in both the insert and vector. With the development of high-fidelity DNA polymerases and PCR technology, many restriction/ligation-independent cloning techniques have been presented and widely used. By using numerous carefully designed primers, manmade cohesive sites can be introduced in some seamless cloning methods, such as enzyme-free cloning [Citation1], hetero-stagger cloning [Citation2] and polymerase incomplete primer extension (PIPE) cloning [Citation3,Citation4]. Consequently, DNA fragments and linear vectors can be paired using the manmade cohesive termini, and the incision sites can be repaired in vivo after direct transformation into Escherichia coli. In addition, a cohesive terminus with 12 bases can be introduced in uracil DNA-glycosylase cloning (UDG) [Citation5,Citation6] and ligation-independent cloning (LIC) [Citation7]; these cloning strategies take advantage of the 3′-5′ exonuclease activities of uracil DNA–glycosylase and T4 DNA polymerase, respectively.
RF cloning [Citation8,Citation9], according to the principle of QuikChange™ site-directed mutagenesis [Citation10,Citation11], provides a convenient, economical and accurate approach for inserting a single fragment into a vector. This method does not introduce any unwanted sequences, such as restriction sites or recombinant sequences. As a derivative method of RF cloning, CPEC cloning [Citation12,Citation13], which is based on the polymerase extension mechanism, extends overlapping regions between the insert and vector fragments to form a complete circular plasmid. However, the amplification of the DNA product in RF cloning and CPEC cloning is linear, which leads to lower yields [Citation14,Citation15]. Using overlap extension PCR [Citation16] and introducing a reverse primer, exponential amplification can be achieved in EMP cloning [Citation14] and IFPC cloning [Citation17]. However, the disadvantage of EMP cloning and IFPC cloning is the use of some indispensable and expensive enzymes (T4 PNK [T4 polynucleotide kinase] and T4 DNA ligase) in the reaction process [Citation15].
Homologous recombination is another widely used cloning method. The use of commercial recombinases, such as Gateway™ (Invitrogen, Carlsbad/CA) [Citation18] and In-Fusion™ (Clontech, Mountain View/CA) [Citation19], can achieve in vitro recombination in homologous regions between the linear insert and the vector. Although this method of homologous recombination is more convenient to manipulate, expensive recombinases are required to complete the entire cloning process. Therefore, several recombinase-free seamless DNA cloning methods using the endogenous homologous recombination activity of laboratory E. coli strains have been reported [Citation20–24]. The Seamless Ligation Cloning Extract (SLiCE) technique uses homologous recombination activities in vitro, in seamless DNA assembly [Citation20–22]. Original SliCE required a specific strain, E. coli PPY, which expresses lambda prophage Red/ET system in E. coli [Citation20]. To extend the utility of this system, the SLiCE preparation method was improved and the cloning efficiency was increased [Citation24]. Improved SLiCE can be prepared from laboratory E. coli strains containing JM109, DH5a and XL10-Gold [Citation24] without any protein expression and has low-cost and versatility for seamless cloning [Citation21]. The in vivo E. coli cloning (iVEC) is an alternative low-cost seamless cloning method [Citation22,Citation23]. The iVEC-method can directly transform and assembly DNA fragments without any treatment to E. coli competent cells, using in vivo E. coli endogenous recombination activities [Citation22,Citation23]. However, the iVEC-method requires high-efficiency E. coli competent cells for efficient seamless cloning [Citation22].
Inspired by the aforementioned cloning methods, a new and effective cloning method was introduced. In this method, based on two parallel overlap extension PCRs, two linear plasmids with the same sequences and different break sites were amplified. After mixing, denaturing and annealing of the two parallel linear plasmids, a manmade cohesive terminus was generated and paired, and the two nicks in the circular plasmid were repaired in vivo in E. coli. Since the new cloning method can generate Manmade Cohesive Termini, we named it MCT cloning. With MCT cloning, the assembly of large DNA fragments (1–4 kbp) can be simply performed in one day. Compared to other seamless cloning methods, MCT cloning is much more efficient and economical for longer fragment insertions.
Materials and methods
Materials
E. coli TOP10F’ (Invitrogen, RecA- strain) was used for cloning. Chemically competent TOP10F’ cells were homemade (approximately 106 cfu/µg). High-fidelity DNA polymerase (KOD-FX) was purchased from TOYOBO (Osaka, Japan). All insert fragments generated from hNav1.5 were inserted into the plasmid pGADT7 (7988 bp, Clontech). All primers were purchased from Sangon (Shanghai, China). The sequences of insert fragments, insertion sites and primers are provided in the Supplementary Tables S1 and S2.
Overview of MCT cloning
The mechanism of MCT cloning for the insertion of single fragments is shown in . MCT cloning requires four steps. The first step is the exponential amplification of the interested fragments (). This amplification process involves two completely independent PCRs, requiring a total of two pairs of primers (F1/R1 and F2/R2 in ). The primer pair F1/R1 and the primer pair F2/R2 were used to amplify the target gene in the first round of PCR, and produce the PCR products M1 and M2, respectively. The reverse primers R1 and R2 contain 3′ regions (the black part in ) homologous to the insert gene and 5′ regions homologous to the vector (the sky blue part in ). The forward primer F1 was homologous to the target genes (the orange part and black part in ), while the forward primer F2 was truncated. Compared with the F1 primer, the forward primer F2 deliberately deleted a partial sequence of the 5′ part (the orange part in ). Therefore, after the first PCR, compared to M1, M2 lacked some of the base pairs (the orange part in ).
Figure 1. Schematic diagram and flowchart of MCT cloning for fragment insertion. The cloning strategy contains four steps: (A) amplification of the insert fragment (gene a) using two pairs of primers (F1/R1 and F2/R2) to get the products M1 and M2; (B) exponential amplification using fragments generated in the first PCR and reverse primers R3 and R4 respectively; (C) digestion by DpnI to eliminate the parent DNA, then mixing, denaturing and annealing; and (D) transformation into chemically competent Top 10F’ cells. Note: The orange and black part represents the target gene; the reddish-purple part represents the vector; the sky blue parts represent the regions homologous to the vector; the orange parts represent the regions homologous to the target gene.
In the second step, the PCR products M1 and M2 were used together with the short reverse primers R3 and R4 to exponentially amplify the vector pGADT7 to get the PCR products M3 and M4, respectively. The reverse primer R3 was homologous to the vector (the reddish-purple part in ), while the 5′ end of the reverse primer R4 added a complementary part of the orange part (the orange part in ), which was deleted in primer F2. Finally, the orange parts were in the 5′ and 3′ ends of M3 and M4, respectively.
In the third step, the methylated templates of the second step were digested with DpnI. After the mixing, denaturing and annealing of M3 and M4, we obtained the final products M5 and M6, in which the orange part sequences were turned into cohesive terminus ().
In the fourth step, the final products M5 and M6 were transformed into chemically competent E. coli Top 10F’ cells, in which the incision sites in the M5 and M6 can be repaired in vivo ().
The first PCR step: amplification of insert fragments
DNA fragments of different sizes (1-4 kbp) were amplified from hNav1.5 using 1 U of KOD-FX enzyme (TOYOBO, Osaka, Japan) supplemented with 0.2 µmol/L of each primer, 1 × reaction buffer, 0.4 mmol/L dNTPs and 10 ng template DNA in 50 µL of the reaction mixture. Mixtures were first denatured (2720 Thermal Cycler, Applied Biosystems, USA) at 94 °C for 2 min, subjected to 25 cycles of denaturation at 98 °C for 10 s, annealing at 60 °C for 30 s and elongation at 68 °C for 25 cycles (1 kbp/min), and followed by a final 10-min extension step at 68 °C. PCR products were analyzed by agarose gel electrophoresis, and target fragments were purified using PCR purification kits (BioSci, Hangzhou, China).
The second PCR step: insertion of fragments
Fragments purified from the first PCR step and the corresponding reverse primer (R3, R4) were used together to exponentially amplify the plasmid pGADT7 (as shown in ). The 50 µL of PCR mixture contained 0.2 µmol/L primer, 10 ng of recipient plasmid, 1 × PCR Buffer, 0.4 mmol/L dNTPs and 1 U KOD-FX polymerase. Mixtures were pre-degenerated at 94 °C for 2 min, subjected to 25 cycles of denaturation at 98 °C for 10 s, annealing at 60 °C for 30 s and elongation at 68 °C (1 kbp/min), and followed by a final 10-min extension step at 68 °C. PCR products were analyzed by agarose gel electrophoresis, and the second PCR products were purified using PCR purification kits or gel extraction kits (BioSci, Hangzhou, China).
The third step: DpnI digestion, mixing, denaturation and annealing of the second PCR products
After purification, the second PCR products were digested using DpnI as follows: purified products (8 µL) supplemented with 1 µL of 10 × reaction buffer were digested with 1 µL of FastDigest™ DpnI (Termo Fermentas, Burlington, Canada) at 37 °C for 1 h. After digestion, the two products were mixed (M3 and M4 in ) in equal amounts(Unless otherwise stated, the dosage of DNA transformed to competent cells in this study was 50 ng for each PCR product). Mixtures were denatured at 98 °C for 30 min and annealed at 37 °C for 20 min.
The fourth step: transformation, colony PCR and DNA sequencing
The products of the third step were transformed directly into 50 µL of chemically competent TOP10F’ cells. All cells were spread onto Luria–Bertani (LB) plates containing 100 µg/mL of ampicillin sodium salt and incubated overnight at 37 °C. Colony numbers per plate were determined to estimate the cloning efficiencies. Colony PCR was used to screen 24 colonies randomly picked from each group. The percentages of positive clones obtained via colony PCR were used to estimate cloning fidelity. Colony PCR was performed using DreamTaq DNA polymerase (Thermo Scientific, USA) according to the manufacturer’s instructions. Further DNA sequencing was performed by Sangon (Shanghai, China).
Results and discussion
Efficiency and fidelity of MCT cloning
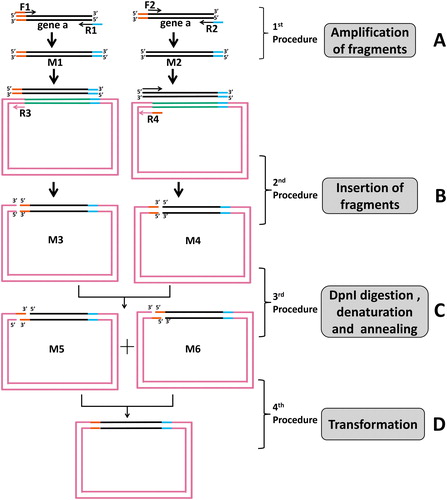
To confirm the feasibility of the MCT cloning method, we first attempted to insert a fragment (1 kbp) into pGADT7 (7988 bp) with different gap sequence sizes. In the first PCR, we used six pairs of primers to amplify the interesting fragment and obtained two PCR products of A (, lane 1) and B1 to B5 (, lanes 1–6). The product A was the complete gene of the insert, while the 5′ ends of B1 to B5 were lacking 10, 15, 20, 25 and 30 bp, respectively. In the second PCR, the first PCR products A, B1, B2, B3, B4 and B5 were used as primers for pGADT7 amplification with the corresponding reverse primers. The second PCR products of A′, B1′, B2′, B3′, B4′ and B5′ were purified using PCR purification kits (, lanes 1–6) and gel extraction kits (, lanes 1–6). After the second PCR, we obtained two linear PCR products with different termini (such as M3 and M4 in ), then we denatured the two products and mixed them in equal amounts to denature and anneal them further to get the two circular plasmids (such as M5 and M6 in ) with a manmade cohesive termini, which could be complemented and repaired in vivo in E. coli. After transformation, more colonies (>400 cfu with 25 bp and 30 bp of cohesive terminus) were obtained with a longer cohesive terminus, while the positive efficiencies were all high (all approximately 100%) ( and ). Interestingly, compared to the other cohesive terminus region length groups, the group with the 10 bp of cohesive terminus yielded enough colonies (>200) and a positive percentage (approximately 100%). Furthermore, for the clear bands of inserts after the second PCR, the procedure of gel extraction did not help to increase the number of colonies and positive percentages ( and ).
Figure 2. Efficiency and fidelity of MCT cloning for different gap sequence sizes, purification methods and dosage of second PCR products. (A) The first PCR of insert-1 (1 kbp, Supplementary Table S1). For lanes 1–6, insert-1 was amplified from hNav1.5 with primers P1/P7, P2/P8, P3/P8, P4/P8, P5/P8 and P6/P8 (Supplementary Table S2), respectively, to produce complete gene and genes lacking 10, 15, 20, 25 and 30 bp, respectively. The arrow indicates the target bands of the first PCR. (B) and (C) For lanes 1–6, the pGADT7 vector was amplified by the first PCR products (lanes 1–6 in , respectively) with reverse primers P9-P14 (Supplementary Table S2), respectively. The fragments were purified by PCR purification kits or by gel extraction kits. The arrow indicates the target band of the second PCR products. (D) and (E) Colony numbers and percentage of positive clones per plate were counted to estimate the cloning efficiencies and fidelities for different sizes of gap sequences, respectively. (F) and (G) Colony numbers and positive clones per plate were counted to estimate the cloning efficiencies and fidelities for different dosages of the second PCR products, respectively. The results are mean values ± SEM of three independent experiments. M: DNA molecular weight marker.
We then examined the effect of the dosage of the second PCR products for cloning efficiency and fidelity. A series tests were run using equal amounts of M5 and M6 (10, 25, 50 and 100 ng each) of the second PCR products as shown in and . With the increase in the second PCR products, the number of recombinant colonies was normally distributed in the bell-shaped curve (). When the amounts were less than 50 ng for each product, M5 and M6, the number of recombinant colonies increased (). However, the number of recombinant colonies decreased when the amounts were more than 50 ng for each product, M5 and M6 (). Despite this, the positive percentage may have changed very little (>90%) ().
Insertion of long fragments of different sizes
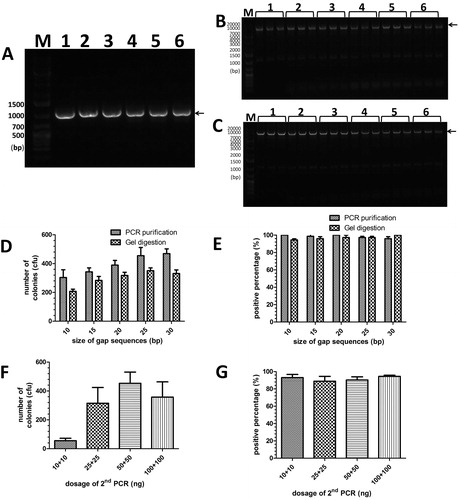
To verify whether the cloning method has a wider applicability, a series of fragments (1–4 kbp) were tested by inserting them into the pGADT7 vector. According to the previous experimental results, we used a 10 bp gap sequence length in this group of experiments (). The 1–3 kbp of insert fragments M1 and M2 were clearly amplified in the first PCR () and produced the products M3 (, lanes 1, 3 and 5) and M4 (, lanes 2, 4 and 6) as primers in the second PCR. For the 1-kb and 2-kb inserts, 10 bp of cohesive sequence produced enough recombinant colonies (>100) and high positive percentages (close to 100%). In contrast, for the inserts longer than 2 kbp (3 kbp, in ), recombinant colonies of the groups with 10 bp of cohesive terminus dropped (below 100 cfu), and the positive percentage decreased to only approximately 20%, although this was partly due to the poor amplification in the second PCR (, lanes 5 and 6).
Figure 3. Insertion of 1–3 kbp of fragments by MCT cloning. (A) For lanes 1–6, insert-1, insert-2 and insert-3(Supplementary Table S1, 1 kbp, 2kbp and 3kbp, respectively) was amplified by the primers P1/P7 and P2/P8, P1/P15 and P2/P16, and P1/P17 and P2/P18 (Supplementary Table S2), respectively. The arrow indicates the target band of the first PCR products. (B) The pGADT7 vector was amplified by the first PCR products (lanes 1–6 in , respectively) with respective reverse primers (P9 for lanes 1, 3 and 5, and P10 for lanes 2, 4 and 6, Supplementary Table S2). The arrow indicates the target band of the first PCR products. (C) and (D) Colony numbers and percentage of positive clones per plate were counted to estimate the cloning efficiencies and fidelities for various lengths of large fragments (1 kbp, 2 kbp and 3 kbp) with 10 bp of gap sequences. The results are mean values ± SEM of two independent experiments. M: DNA molecular weight marker.
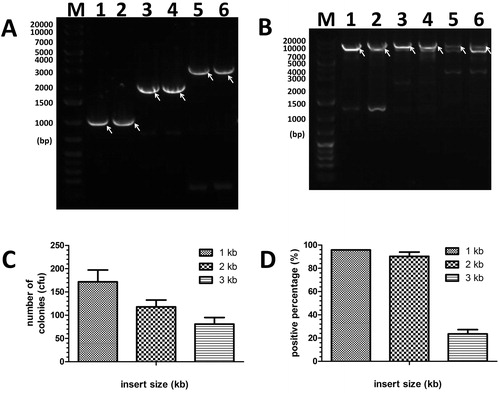
Therefore, longer cohesive sequences (30 bp) were tested to overcome the limitation, as shown in . The insert fragments M1 and M2 of 3 kbp and 4 kbp were clearly amplified in the first PCR () and amplified the corresponding products M3 and M4 () as primers in the second PCR, respectively. With the longer cohesive terminus, the number of recombinant colonies of groups of 3 kbp and 4 kbp inserts was increased (), and the positive percentages improved up to approximately 50% (), although the second PCR products M5 and M6 of 3 kbp and 4 kbp inserts were still poorly amplified with non-specific bands.
Figure 4. Insertion of 3–4 kbp of fragments by MCT cloning. (A) For lanes 1–4, insert-3 and insert-4 (Supplementary Table S1, 3 kbp and 4kbp, respectively) was amplified by the primers P1/P17 and P6/P18, and P1/P19 and P6/P20 (Supplementary Table S2), respectively. The arrow indicates the target band of the first PCR. (B) The purified fragments (3 kbp and 4 kbp [lanes 1–2 and lanes 3–4]) from the 1st PCR were inserted into pGADT7 (7988 bp). The arrow indicates the target band of the second PCR. (C) and (D) Colony numbers and percentage of positive clones per plate were counted to estimate the cloning efficiencies and fidelities for various lengths of large fragments (3 kbp and 4 kbp) with 30 bp of gap sequences. The results are mean values ± SEM of two independent experiments. M: DNA molecular weight marker.
![Figure 4. Insertion of 3–4 kbp of fragments by MCT cloning. (A) For lanes 1–4, insert-3 and insert-4 (Supplementary Table S1, 3 kbp and 4kbp, respectively) was amplified by the primers P1/P17 and P6/P18, and P1/P19 and P6/P20 (Supplementary Table S2), respectively. The arrow indicates the target band of the first PCR. (B) The purified fragments (3 kbp and 4 kbp [lanes 1–2 and lanes 3–4]) from the 1st PCR were inserted into pGADT7 (7988 bp). The arrow indicates the target band of the second PCR. (C) and (D) Colony numbers and percentage of positive clones per plate were counted to estimate the cloning efficiencies and fidelities for various lengths of large fragments (3 kbp and 4 kbp) with 30 bp of gap sequences. The results are mean values ± SEM of two independent experiments. M: DNA molecular weight marker.](/cms/asset/de0aa7e9-18d9-432a-8e6a-e5687f3bf702/tbeq_a_1507756_f0004_b.jpg)
In the common procedure of gene cloning (< 3000 bp, approximately 1000 amino acids), the MCT cloning method can obtain enough colonies and a positive percentage under low chemical transformation level (about 106 cfu/µg). Similar strategies of MCT cloning have been reported recently, such as TPA cloning [Citation25] and MRF cloning [Citation26], which also use two groups of primers to make manmade cohesive termini. However, the manmade cohesive termini are generated in both ends of the inserts and the vector. The nicked circular plasmid with multiple gaps should be repaired by using T4 DNA ligase (MRF cloning) or chemically competent cells with high efficiency (about 5 × 107 cfu/µg in TPA cloning). In contrast, only one gap is generated in the MCT cloning by using overlap PCR, which could generate more positive products by using home-made competent cells with low transformation efficiency (about 106 cfu/µg). In summary, from the operational point of view, the MCT cloning method, like the iVEC method [Citation23], in which the entire cloning process including DNA amplification, DpnI digestion and transformation can be completed within 1 to 2 days, is a simple and rapid DNA assembly technique. In addition, in terms of experimental cost, the MCT cloning method, like SLiCE cloning [Citation21,Citation22], which needs home-made competent E. coli cells, is more economical than iVEC [Citation23], which requires commercially high-efficiency competent E. coli cells.
Conclusions
Here, we developed a novel cloning method that provides an alternative approach to inserting DNA fragments into plasmids. This new method accomplished large fragment (1–4 kbp) assembly with high efficiency and high fidelity. MCT cloning, as a derivative of RF cloning, and its derivatives (CPEC cloning, EMP cloning and IFPC cloning) offer the advantage of both low cost (RF cloning and CPEC cloning) and exponential amplification (EMP cloning and IFPC cloning). We expect MCT cloning to play a significant role in functional genomic studies, protein engineering and other related processes.
| Abbreviations | ||
| PIPE | = | incomplete primer extension |
| UDG | = | uracil DNA-glycosylase |
| LIC | = | ligation-independent cloning |
| RF | = | restriction-free |
| CPEC | = | circular polymerase extension cloning |
| EMP | = | exponential megapriming PCR |
| IFPC | = | inverse fusion PCR cloning |
Disclosure statement
The authors declare they have no competing interests.
Additional information
Funding
References
- Tillett D, Neilan BA. Enzyme-free cloning: a rapid method to clone PCR products independent of vector restriction enzyme sites. Nucleic Acids Res. 1999;27:e26–e28.
- Liu Z. Hetero-stagger cloning: efficient and rapid cloning of PCR products. Nucleic Acids Res. 1996;24:2458–2459.
- Klock HE, Koesema EJ, Knuth MW, et al. Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins. 2008;71:982–994.
- Klock HE, Lesley SA. The Polymerase Incomplete Primer Extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. Methods Mol Biol. 2009;498:91–103.
- Rashtchian A, Buchman GW, Schuster DM, et al. Uracil DNA glycosylase-mediated cloning of polymerase chain reaction-amplified DNA: application to genomic and cDNA cloning. Anal Biochem. 1992;206:91–97.
- Nisson PE, Rashtchian A, Watkins PC. Rapid and efficient cloning of Alu-PCR products using uracil DNA glycosylase. PCR Methods Appl. 1991;1:120–123.
- Aslanidis C, de Jong PJ. Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 1990;18:6069–6074.
- van den Ent F, Lowe J. RF cloning: a restriction-free method for inserting target genes into plasmids. J Biochem Biophys Methods. 2006;67:67–74.
- Unger T, Jacobovitch Y, Dantes A,et al. Applications of the Restriction Free (RF) cloning procedure for molecular manipulations and protein expression. J Struct Biol. 2010;172:34–44.
- Braman J, Papworth C, Greener A. Site-directed mutagenesis using double-stranded plasmid DNA templates. In: Trower MK, editor. In vitro mutagenesis protocols. Totowa (NJ): Humana Press; 1996. p. 31–44.
- Bauer JC (SD, CA), Wright DA (Cambridge, MA), Braman JC (Carlsbad, CA), et al., inventors; Stratagene (La Jolla, CA), Children's Medical Center Corp. (Longwood, MA), assignee. Circular site-directed mutagenesis. United States Patent 6391548. 2002.
- Quan J, Tian J. Circular polymerase extension cloning of complex gene libraries and pathways. PLoS One. 2009 [cited 2009 Jan 16];4:e6441. [6 p.] DOI:10.1371/journal.pone.0006441
- Quan J, Tian J. Circular polymerase extension cloning for high-throughput cloning of complex and combinatorial DNA libraries. Nat Protoc. 2011;6:242–251.
- Ulrich A, Andersen KR, Schwartz TU. Exponential megapriming PCR (EMP) cloning–seamless DNA insertion into any target plasmid without sequence constraints. PLoS One. 2012 [cited 2012 Dec 31];7:e53360. [9 p.] DOI:10.1371/journal.pone.0053360
- Liu C-J, Jiang H, Wu L, et al OEPR cloning: an efficient and seamless cloning strategy for large- and multi-fragments. Sci Rep-UK. 2017 [cited 2017 Mar 16];7:44648 [p7] DOI:10.1038/srep44648
- Ho SN, Hunt HD, Horton RM,et al. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59.
- Spiliotis M. Inverse fusion PCR cloning. PloS One. 2012 [cited 2017 Mar 16];7:e35407. [8 p.] DOI:10.1371/journal.pone.0035407
- Karimi M, Inzé D, Depicker A. GATEWAY™ vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci. 2002;7:193–195.
- Zhu B, Cai G, Hall EO, et al. In-fusion assembly: seamless engineering of multidomain fusion proteins, modular vectors, and mutations. BioTechniques. 2007;43:354–359.
- Zhang Y, Werling U, Edelmann W. SLiCE: a novel bacterial cell extract-based DNA cloning method. Nucleic Acids Res. 2012 [cited 2012 Apr 1];40:e55 [p 10] DOI:10.1093/nar/gkr1288
- Okegawa Y, Motohashi K. A simple and ultra-low cost homemade seamless ligation cloning extract (SLiCE) as an alternative to a commercially available seamless DNA cloning kit. Biochem Biophys Rep. 2015;4:148–151.
- Motohashi K. Evaluation of the efficiency and utility of recombinant enzyme-free seamless DNA cloning methods. Biochem Biophys Rep. 2017;9:310–315.
- Kostylev M, Otwell AE, Richardson RE, et al. Cloning should be simple: Escherichia coli DH5α-mediated assembly of multiple DNA fragments with short end homologies. PloS One. 2015 [cited 2015 Sep 5];10:e0137466 [p 15] DOI:10.1371/journal.pone.0137466
- Motohashi K. A simple and efficient seamless DNA cloning method using SLiCE from Escherichia coli laboratory strains and its application to SLiP site-directed mutagenesis. BMC Biotechnol. 2015 [cited 2015 June 3];15(1):47. [p 9]DOI:10.1186/s12896-015-0162-8
- Liang J, Liu Z, Low XZ, et al. Twin-primer non-enzymatic DNA assembly: an efficient and accurate multi-part DNA assembly method. Nucleic Acids Res. 2017 [cited 2017 June 20];45:e94.[p 11] DOI:10.1093/nar/gkx132
- Zeng F, Hao Z, Li P, et al. A restriction-free method for gene reconstitution using two single-primer PCRs in parallel to generate compatible cohesive ends. BMC Biotechnol. 2017 [cited 2017 Mar 1];17:32.[p 7] DOI:10.1186/s12896-017-0346-5