
Abstract
Early infantile epileptic encephalopathy (EIEE) is a disorder with variable genetic heterogeneity. Symptoms are mostly presented with generalised epileptic seizures with an infantile onset and progressive neurodevelopmental delay. Early infantile epileptic encephalopathy13 is caused by mutations in the SCN8A gene, which encodes the neuronal voltage-gated sodium channel α subunit (Nav1.6) and plays a major role in neuronal excitability. Describing the wide clinical variability of previously reported cases of patients carrying the same mutation, we demonstrate the complexity of the disease and the necessity of correctly correlating the phenotype with the genotype. Here, we present a minireview and a case report of EIEE13 involving the rare p.Arg1872Gln mutation in the SCN8A gene. We used targeted next-generation sequencing to examine a six-year-old girl with complex partial seizures from the left temporal lobe since 4 months of age. The condition was difficult to control with medication and the seizures evolved to generalised tonic-clonic seizures after the age of 3 years. Neurodevelopment in the child became severely delayed although seizures were as rare as 1 in every 5–10 months. А heterozygous missense mutation in the SCN8A gene (NM_014191.3:c.5616G > A, NP_055006.1:p.Arg1872Gln) was found. The variant was validated by Sanger sequencing. We suggest that this SCN8A mutation has a primary neurodegenerative effect leading to brain atrophy and intellectual disability (with or without autism) that is partially independent of its epileptogenic effect. Our results demonstrate that the application of large panels with clinically-associated genes is essential for identifying rare mutations in individuals with disorders of unknown etiology.
Introduction
Early infantile epileptic encephalopathy (EIEE) is a devastating neurologic disorder with heterogeneous aetiology [Citation1, Citation2]. The disease affects new-borns and infants usually within the first 3 months of life presenting with polymorphic generalised or lateralised epileptic seizures resistant to treatment. They are combined with psychomotor deficits and cognitive disabilities, frequently severe, and carry a high risk of sudden unexplained death in epilepsy (SUDEP) due to complications of the disease [Citation2, Citation3]. Diagnosis of EIEEs can be very challenging due to their large phenotypic variability [Citation4]. Some of them comply with the clinico-electroencephalographic criteria of Ohtahara or West syndrome but others lack specificity [Citation5].
Recently it was recognized that EIEE type 13 (EIEE13) is caused by de novo gain-of-function missense mutations in the SCN8A gene. SCN8A is part of the voltage-gated sodium channels (VGSCs) gene family [Citation4, Citation6, Citation7]. Voltage-gated sodium channels are transmembrane proteins with a fundamental role in the initiation and propagation of the action potential in most excitable cells [Citation7–12]. They are composed of nine pore-forming α subunits (260 kDa) and one or two β subunits (30–40 kDa). The α subunit (Nav1.1–Nav1.9) contains four homologous domains (I–IV), each one with a motif of six transmembrane segments (S1–S6), connected with two β subunits [Citation7–9, Citation13–15]. Nine mammalian genes (SCN1A, SCN2A, SCN3A, SCN4A, SCN5A, SCN8A, SCN9A, SCN10A and SCN11A) encode the pore-forming α subunits, and four genes (SCN1B to SCN4B) encode the β subunits. The majority are expressed in the central and peripheral nervous system (SCN1A, SCN2A, SCN3A and SCN8A). SCN9A, SCN10A and SCN1A are expressed only in the peripheral nervous system. SCN4A is highly expressed in the skeletal muscles and SCN5A in the cardiac tissue [Citation11, Citation16].
The Nav1.6 channel (sodium channel, voltage-gated, type VIII, alpha subunit and OMIM *70602) is concentrated at the nodes of Ranvier, the dendrites and the initial segment of the axons [Citation8, Citation9, Citation11]. It is encoded by the SCN8A gene located at human chromosome 12q13.13 [Citation17]. Its coding sequence (170 Kb) contains 27 exons encoding a protein of 1980 amino acid residues intolerant to variations [Citation17–19]. Even a small deviation in the properties of the ion channel proteins can lead to significant functional defects [Citation11].
SCN8A-encephalopathies are autosomal dominant disorders arising from de novo missense mutations in the SCN8A gene [Citation1, Citation19]. The first case of a de novo heterozygous mutation in SCN8A (p.Asn1768Asp) in a child with EIEE13 (EIEE13, OMIM 614558) was described by Veeramah et al. in 2012 [Citation3]. Since then, more than 150 SCN8A de novo mutations have been detected and correlated to the phenotypic spectrum of EIEE13 [Citation4, Citation9, Citation16, Citation18, Citation20–25]. In this review, we give a brief overview of previously reported cases with p.Arg1872Gln mutation in the SCN8A gene and describe the clinical and genetic features of a sporadic case of EIEE13 studied by targeted massive parallel DNA sequencing.
SCN8A p.Arg1872Gln mutation in EIEE13
Literature overview
Clinical diagnosis and treatment of patients with epileptic encephalopathy are challenging, due to the wide variability of symptoms and drug response observed even among individuals sharing one and the same gene mutation () [Citation1, Citation2, Citation26–28]. So far, reported patients carrying the p.Arg1872Gln mutation are characterized with a wide range of phenotypic complexity and differences. Neonatal seizures evolve to different types, developmental delay from normal to moderate and severe. The patients have different electroencephalogram (EEG)/magnetic resonance imaging (MRI) status and broad variability of treatment and drug response [Citation1, Citation2, Citation26–28]. Detailed clinical features of reported cases are described in .
Table 1. Clinical features of published cases with the p.Arg1872Gln mutation in SCN8A.
For precision phenotyping, it is necessary to understand the wide clinical spectrum and associate it with the genotype of the patient. According to Wagnon et al. [Citation2], differences in the genotype, like mutations in other channel genes and/or stochastic variations during nervous system development, are the basis for the phenotype differences between patients with identical mutations.
Clinical report
Here, we present a case of a girl born after a pathological first pregnancy at 37 weeks of gestation. She had mild perinatal asphyxia and a birth weight of 2300 g. Her development was uneventful until the age of 4 months when cyanosis and trembling of the extremities were noticed with a frequency of 2–6 per day during a mild respiratory infection. The EEG, brain computed tomography (CT) and cerebrospinal fluid analysis were normal initially. The seizures were resistant to treatment with phenobarbital and valproate, but reduced in number by oxcarbazepine and clonazepam. Trembling of the lips and vocalization were added to the seizure semiology and the EEG became slow and with few occipital, fronto-central or temporal, predominantly left-sided sharp-and-slow-wave complexes at 7 months of age. A reduction of seizure frequency to 1 in 6 months lasted until the age of 3 years, when a few days after a mild car accident a bout of several prolonged generalised tonic-clonic convulsions appeared, necessitating admission to an intensive care unit. The seizures were stopped by administration of valproic acid. In the next years, such convulsions were rare: 1 in every 5–10 months, the patient being on treatment with valproate and oxcarbazepine until the age of 5 years, and valproate and lamotrigine in the next years. Levetiracetam and carbamazepine were also tried but were inefficacious. Electroencephalograms showed multifocal bilateral epileptiform discharges mostly in the left temporal leads on a slow background (). Brain MRI at 1 year displayed mild cortical atrophy, which was found increased in the left temporal lobe at 3 years (). Blood and urine metabolic tests gave normal results. Despite the absence of frequent seizures, her neurodevelopment gradually delayed, especially in speech, and autistic features became evident after the age of 3 years. At the time of writing, the patient is 8 years old, walks independently on a broad base, speaks about 10 words, sometimes obeys simple commands, can feed herself and is toilet-trained during daytime; her joined attention is reduced, and repetitive and restrictive behaviours are frequent. Neurological and somatic examinations are unremarkable.
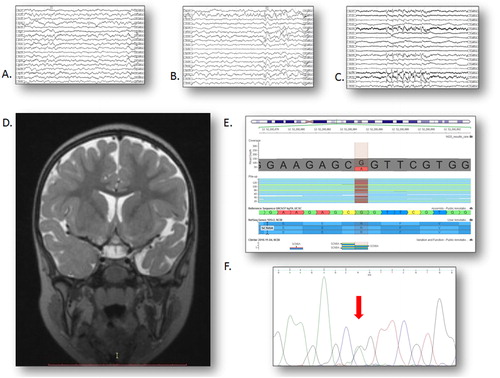
Figure 1. An overview of the neurological and molecular genetic status of the patient. (A) Interictal sleep EEG at 2 years, revealing sharp and slow waves predominantly in the left temporal leads (speed 15 mm/s). (B) Interictal sleep EEG at 3 years, revealing a generalised spike-wave discharge as well as frequent sharp and slow waves predominantly in the left anterior temporal leads (speed 15 mm/s). (C) Interictal sleep EEG at 5 years with a 5-second paroxysm of primary generalised slow spike-and-wave complexes predominant in the anterior leads (speed 15 mm/s). (D) Coronal T2 MRI scan at 3 years, revealing mild cortical atrophy with a more severe affection of the left temporal lobe. (E) NM_014191.3:c.5616G > A variant in exon 27 of the SCN8A gene in our patient; the replaced nucleotide is marked in red. (F) Sanger sequencing electropherogram of the same region.
Methods
After obtaining written informed consent from the parents, genomic DNA was extracted from blood using the standard QIAamp DNA Blood Mini Kit (Qiagen). Next-generation sequencing (NGS) libraries were prepared using the TruSight One gene panel, which provides comprehensive coverage of >4800 disease-associated genes. The library was sequenced on an Illumina MiSeq system (Illumina, USA) with 150-bp paired-end reads, according to the manufacturer protocols. A mean region coverage depth of 103.5× was achieved with 77.3% of target coverage at 50×. Data analysis and variant annotation were performed using the Illumina Isis 2.6.2.3, BWA 0.7.9a, SAM tools 0.1.18, GATK v1.6 and Variant Studio v2.2 software and GenomeBrowse v2.1.1 was used for data visualisation. Our analysis was focused on rare variants (global MAF <1%), in protein-coding regions and exon–intron boundaries of target genes.
Sanger sequencing was performed for validation of the NGS finding. Polymerase chain reaction (PCR) was performed with specific primers designed using the open-access tool Primer3 (http://http://bioinfo.ut.ee/primer3-0.4.0/). Amplified products were run on an ABI Prism 3130xl Genetic Analyzer (Applied Biosystems, USA) according to the manufacturer’s protocols. Primers and ` conditions are available upon request.
DNA samples from the parents were not available for testing.
Results and discussion
A patient with complex clinical features was examined by targeted NGS. After variant filtering and annotation, we identified a rare heterozygous missense mutation in exon 27 of the SCN8A gene: NM_014191.3:c.5616G > A, NP_055006.1: p.Arg1872Gln (). It represents a nucleotide substitution of guanine with adenine at position 52200885 on chromosome 12. The change affects the coding sequence and results in an amino acid substitution from arginine to glutamine at position 1872 of the protein, which is reported as a mutational hotspot [Citation2]. The reading depth at this position was 95×. The SCN8A mutation was successfully confirmed by Sanger sequencing ().
Pathogenic variants in SCN8A are phenotypically expressed in an autosomal dominant manner in EIEE13 and thus affected individuals are heterozygous. The detected variant is not published in the Human Gene Mutation Database, but has been observed to arise de novo in other patients diagnosed with EIEE13 and development delay [Citation1, Citation2, Citation26–28].
Clinically, our patient’s phenotype exemplifies some typical features of EIEE13. The age of onset of seizures complies with the period between 3 and 7 months of age reported for most cases [Citation1, Citation2, Citation26, Citation27]. Seizures were difficult to control with medications as in all cases [Citation1, Citation2, Citation23, Citation26–28]. In most patients, a regression in neurodevelopment was reported, frequently with autistic features which result in intellectual disability [Citation1, Citation2, Citation26, Citation27]. Electroencephalogram might be normal initially but epileptiform activity, mostly generalized, becomes more evident with age and accompanied by background slowing and brain atrophy [Citation1, Citation2, Citation26–28].
The present case displays some peculiarities, as well:
Unlike other EIEE13 patients in whom infantile spasms or other primary generalized seizures dominate [Citation1, Citation2, Citation23], our case, in the first 3 years of life, presents with semiology, interictal EEGs and MRI, suggesting complex partial seizures originating from the left temporal lobe. Only after that age did the seizures become primary generalized tonic-clonic.
Unlike other EIEE13 patients who display polymorpic seizures [Citation1, Citation2, Citation9, Citation19], our patient had one type of seizure in the first 3 years that evolved to a second type afterwards.
Although medication-resistant, the seizures in our patient are rare, i.e., one in several months.
The concept of epileptic encephalopathy is based on the assumption that frequent seizures and subclinical epileptiform activity cause the encephalopathy [Citation5]. In the present case, the neurodevelopmental delay does not coincide with frequent seizures. On the contrary, despite seizure rarity (1 in 10 months) and absence of marked interictal epileptiform activity, the neurodevelopment of our patient became progressively delayed. Psychomotor regression as a major sign of EIEE13 is pointed out by other authors as well [Citation2, Citation9, Citation23, Citation26, Citation27]. Progressive brain atrophy parallels neurodevelopmental delay [Citation2, Citation9, Citation19, Citation27, Citation28] and may even lead to microcephaly [Citation23, Citation27]. The semiological, EEG and MRI data imply temporal lobe epilepsy, especially in the first years of life. Cortical dysplasias were meticulously searched for and ruled out. There were no history data of febrile seizures and no hippocampal sclerosis on MRI. The discrepancy between the rarity of seizures and the more severe neurodevelopmental delay suggested the possibility of a genetic encephalopathy and urged the genetic studies. All these data suggest that the mutations in SCN8A have a primary neurodegenerative effect, besides the epileptogenic effect.
The rare known missence mutation (c.5616G > A, p.Arg1872Gln) in SCN8A detected in our patient is already a subject of interest for other authors [Citation1, Citation2, Citation26–28]. According to several groups, the amino acid substitution (Arg1872Gln) was predicted to be extremely damaging to the structure of the channel [Citation9, Citation29–32]. The arginine residue on position 1872 is highly conserved, located in the cytoplasmic C-terminal domain of the Nav1.6 channel. Through cooperation between positively and negatively charged residues, Arg1872 interacts with the inactivation gate of Nav1.6 [Citation2, Citation33]. As a result of the mutation, the Nav1.6 channel inactivation is impaired by the loss of the positive charge of the arginine residue 1872 [Citation2].
Wagnon et al. [Citation2] in 2016 demonstrated the functional effects of the mutation in arginine 1872 by doing in vitro functional analysis. They introduced the mutation (Arg1872Gln) into the full-length Nav1.6 cDNA transfected into the neuron-derived cell line ND7/23. The results proved that the Nav1.6 channel of the mutant cells were impaired in transition from open to inactivated state resulting in channel hyperactivity associated with gain-of-function [Citation2, Citation30, Citation31, Citation34, Citation35]. The biophysical properties of the mutant channel prove the role of SCN8A as a critical mediator of neuronal excitability, revealing the relationship between the function-structure characteristics of the Nav1.6 channel and the pathogenesis of epilepsy and intellectual disability [Citation16, Citation18].
In recent years, more than 150 mutations in the SCN8A gene have been detected through NGS in patients with epilepsy and developmental delay [Citation18]. Approximately 1% of the patients with unexplained infantile-onset epileptic encephalopathy are carriers of a mutation in SCN8A [Citation1].
Conclusions
Our finding demonstrates the usefulness of NGS in clinical neurology and the wide range of applications of the TruSight One gene panel, to successfully detect rare mutations in sporadic cases of epileptic encephalopathy of unknown aetiology. Such studies provide more insight into the pathogenesis of the disease as well as allow a personalised approach and treatment and could be useful as a reference for further studies.
Disclosure statement
The authors report no conflict of interest. The authors alone are responsible for the content and writing of this article.
References
- Larsen J, Carvill GL, Gardella E. The phenotypic spectrum of SCN8A encephalopathy. Neurology. 2015;84:480–489.
- Wagnon JL, Barker BS, Hounshel JA, et al. Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Ann Clin Transl Neurol. 2016;3:114–123.
- Veeramah KR, O’Brien JE, Meisler MH, et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet. 2012;90:205–510.
- Carvill LG, Heavin SB, Yendle SC, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;7:825–831.
- Nordli DR. Jr. Epileptic encephalopathies in infants and children. J Clin Neurophysiol. 2012;29:420–424.
- Mantegazza M, Rusconi R, Cestèle S. Mutations of ion channels in genetic epilepsies. In: Striano P, editor. Epilepsy towards the next decade - new trends and hopes in epileptology. Switzerland: Springer; 2015. p. 15–34.
- de Lera Ruiz M, Kraus RL. Voltage-gated sodium channels: structure, function, pharmacology, and clinical indications. J Med Chem. 2015;58:7093–7118.
- Oliva M, Berkovic SF, Petrou S. Sodium channels and the neurobiology of epilepsy. Epilepsia. 2012;53:1849–1859.
- Ohba C, Kato M, Takahashi S, et al. Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia. 2014;55:994–1000.
- George AL. Jr. Inherited disorders of voltage-gated sodium channels. J Clin Invest. 2005;115:1990–1999.
- Meisler MH, Kearney JA. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest. 2005;115:2010–2017.
- Makinson CD, Tanaka BS, Sorokin JM, et al. Understanding network connections connects genotype to epilepsy phenotype. Neuron. 2017;93:1165–1179.
- Huang W, Liu M, Yan SF, et al. Structure-based assessment of disease related mutations in human voltage-gated sodium channels. Protein Cell. 2017;8:401–438.
- Namadurai S, Yereddi NR, Cusdin FS, et al. A new look at sodium channel β subunits. Open Biol. 2015;5:140192. DOI: 10.1098/rsob.140192
- Brackenbury WJ, Isom LL. Na Channel β Subunits: Overachievers of the Ion Channel Family. Front Pharmacol. 2011;2:53. [cited 2018 Mar 04]DOI: 10.3389/fphar.2011.00053
- Blanchard MG, Willemsen MH, Walker JB, et al. De novo gain-of-function and loss-of-function mutations of SCN8A in patients with intellectual disabilities and epilepsy. J Med Genet. 2015;52:330–337.
- Plummer NW, Galt J, Jones JM, et al. Exon organization, coding sequence, physical mapping, and polymorphic intragenic markers for the human neuronal sodium channel gene SCN8A. Genomics. 1998;54:287–296.
- O’Brien JE, Meisler MH. Sodium channel SCN8A (Nav1.6): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Front Genet. 2013;4:213. DOI: 10.3389/fgene.2013.00213
- Wagnon JL, Meisler MH. Recurrent and non-recurrent mutations of SCN8A in epileptic encephalopathy. Front Neurol. 2015;6:104. DOI: 10.3389/fneur,2015.00104
- Wagnon JL, Barker BS, Ottolini M, et al. Loss-of-function variants of SCN8A in intellectual disability without seizures. Neurol Genet. 2017;3:e170. [cited 2018 Mar 04]DOI: 10.1212/NXG.0000000000000170
- Meisler MH, Helman G, Hammer MF, et al. SCN8A encephalopathy: Research progress and prospects. Epilepsia. 2016;57:1027–1035.
- Vaher U, Noukas M, Nikopensius T, et al. De Novo SCN8A mutation identified by whole-exome sequencing in a boy with neonatal epileptic encephalopathy, multiple congenital anomalies, and movement disorders. J Child Neurol. 2014;29:NP202–NP206.
- de Kovel CG, Meisler MH, Brilstra EH, et al. Characterization of a de novo SCN8A mutation in a patient with epileptic encephalopathy. Epilepsy Res. 2014;108:1511–1518.
- Estacion M, O'Brien JE, Conravey A, et al. A novel de novo mutation of SCN8A (Nav1.6) with enhanced channel activation in a child with epileptic encephalopathy. Neurobiol Dis. 2014;69:117–123.
- Wang J, Gao H, Bao H, et al. SCN8A mutations in Chinese patients with early onset epileptic encephalopathy and benign infantile seizures. BMC Med Genet. 2017;18:104. DOI: 10.1186/s12881-017-0460-1
- Arafat A, Jing P, Ma Y, et al. Unexplained early infantile epileptic encephalopathy in Han Chinese children: next-generation sequencing and phenotype enriching. Sci Rep. 2017;7:46227. DOI: 10.1038/srep46227
- Horvath GA, Demos M, Shyr C, et al. Secondary neurotransmitter deficiencies in epilepsy caused by voltage-gated sodium channelopathies: A potential treatment target? Mol Genet Metab. 2016;117:42–48.
- Hussain A, Seinfeld S, Morton L. Genetic association with Ictal cardiorespiratory phenomena: SCN8A Case series. J Pediatr Neurol. 2016;14:151–155.
- Eijkelkamp N, Linley JE, Baker MD, et al. Neurological perspectives on voltage-gated sodium channels. Brain. 2012;135:2585–2612.
- Trudeau MM, Dalton JC, Day JW, et al. Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia and mental retardation. J Med Genet. 2006;43:527–530.
- Anand G, Collett-White F, Orsini A, et al. Autosomal dominant SCN8A mutation with an unusually mild phenotype. Eur J Paediatr Neurol. 2016;20:761–765.
- Vacher H, Trimmer JS. Trafficking mechanisms underlying neuronal voltage-gated ion channel localization at the axon initial segment. Epilepsia. 2012;53:21–31.
- Lee A, Goldin AL. Role of the amino and carboxy termini in isoform-specific sodium channel variation. J Physiol (Lond). 2008;586:3917–3926.
- Nguyen HM, Goldin AL. Sodium channel carboxyl-terminal residue regulates fast inactivation. J Biol Chem. 2010;285:9077–9089.
- Orsini A, Zara F, Striano P. Recent advances in epilepsy genetics. Neurosci Lett. 2018;667:4–9.