
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
In the light of the emerging bacterial resistance to broad-spectrum antibiotics, the search for new antibacterial therapeutics and drug combinations is one of the most challenging topics nowadays. In the present study, we investigated for the first time the antibacterial and biofilm inhibitory effects of the third generation anticancer alkylphosphocholine (APC) erufosine against pathogenic Staphylococcus aureus strains in comparison to the prototype of this pharmacological class of drugs, miltefosine. We also searched for synergistic antibacterial combinations between both APCs and curcumin incorporated in copolymeric micelles based on Pluronic® P123 or a mixture of Pluronic® P123 and Pluronic® F127 (P123/F127). The obtained quantitative redox-activity experimental data and drug–drug interactions were evaluated by using mathematical models in the MAPLE software. Similar to miltefosine, erufosine showed a moderate bacteriostatic effect in clinically relevant concentrations (50 ÷ 60 µmol/L) and inhibited the redox activity of the treated bacteria up to 90% at minimal inhibitory concentrations. The effect of both APCs towards methicillin resistant staphylococci was enhanced by combinations with P123/F127 micellar CRM at a ratio of 1:1. Erufosine showed a stronger median biofilm inhibition at lower concentrations (MBIC50 = 1.87 µmol/L) than miltefosine (MBIC50 = 6.0 µmol/L) and curcumin (MBIC50 = 24.84 µmol/L) as demonstrated by quantification of biofilm-bound bacteria. In conclusion, the estimated antibacterial activity of erufosine widens the spectrum of its useful pharmacological effects, which is important for its clinical development. The established synergistic and additive drug combinations could be beneficial for the application of both APCs in cancer therapy, since numerous malignancies are accompanied by bacterial infections.
Introduction
The search for new drugs and frontiers in drug–drug interactions used against pathogenic bacteria is one of the hottest and challenging topics in recent years. In the light of the emerging bacterial resistance to broad-spectrum antibiotics, the efforts of many scientists are focused on the development of new active chemical structures or drug repurposing. Alkylphosphocholines (APC) are a promising class of drugs with pleiotropic pharmacological effects [Citation1–3]. They originate from membrane ether lipids and structurally represent phosphocholine esters of aliphatic long-chain alcohols with amphiphilic chemical properties. This structure allows the formation of lamellar or micellar structures in aqueous solutions by a critical micelle concentration comparable to that of lysolecithin [Citation4,Citation5]. APCs not only demonstrate cytotoxic effects against various types of malignant cells but are also highly effective against parasites such as Leishmania spp. [Citation1,Citation6–10], African Trypanosoma spp. [Citation11], Entamoeba histolytica [Citation12], Acanthamoeba spp., Schistosoma mansoni [Citation13], Plasmodium falciparum [Citation14], Gram-positive pathogenic bacteria [Citation15,Citation16], pathogenic fungi [Citation17,Citation18] and RNA or DNA viruses [Citation19]. The first APC, miltefosine (MLT), was obtained by Berger and Eibl from alkyllysophosphocholines by removing the glycerol backbone [Citation20]. The simultaneous but independent discovery of the antiprotozoal and antineoplastic activities of MLT and related alkylphosphocholine drugs occurred in the early 1980s [Citation21]. First reports of the antiprotozoal activity of phospholipid analogues against Tetrahymena pyriformis [Citation22] and Leishmania donovani [Citation23,Citation24] were published during that period of time in parallel to their development as anti-cancer drugs. Remarkably, very similar molecular modes of action of MLT were identified against both, Leishmania parasites and human cancer cells, linking its activity mainly to apoptosis and disturbance of lipid-dependent cell signalling pathways. After successful clinical trials, MLT was registered for local therapy of cutaneous metastasis from breast cancer and as first and still only oral treatment of all types of leishmaniasis [Citation25,Citation26]. In 2010, it was included in the World Health Organisation (WHO) model List of Essential Medicines [Citation27]. MLT has also demonstrated strong antifungal effect against Candida albicans and varying antibacterial activity against several Gram-positive pathogens. It elicited significant in vitro bactericidal effect against pneumococcal bacteria such as Streptococcus pneumonia due to triggering of the pneumococcal autolysin LytA and subsequent autolysis of the pneumococcal cultures [Citation15]. Unfortunately, its in vivo administration in a murine peritonitis–sepsis model was characterized by unresponsiveness because of inadequate pharmacokinetics and/or extracellular localization of the pneumococcus [Citation28]. It has to be noted that only moderate antibacterial activity was observed against pathogenic Staphylococcus aureus strains and vancomycin resistant Enterococcus spp. Gram-negative bacterial species, such as Escherichia coli and Pseudomonas aeruginosa remained resistant to treatment with MLT. Results similar to those of MLT were also obtained for other APCs [Citation16]. All these findings prompted a series of scientific investigations aiming both, to search for novel antiprotozoal, antifungal, antibacterial and antiviral therapeutics or to reveal the antimicrobial potential of already known APCs’ structures [Citation1,Citation16].
Given these data, it remains very challenging to investigate the new third-generation APC-derivate erufosine (ERF) for antibacterial activity [Citation29,Citation30]. This drug, which is very attractive from the pharmacological point of view, has been developed by extending the aliphatic alcohol chain up to 22 carbon atoms and introducing a ω-9–cis-double bond [Citation31,Citation32]. In contrast to MLT, which forms micellar aqueous solutions and cannot be applied intravenously due to hemolytic properties, ERF forms lamellar aqueous solutions, and is suitable for intravenous administration [Citation4,Citation5,Citation33]. ERF exerts remarkably strong antineoplastic activity in vitro against various types of neoplasias related to bacterial infections, such as oral squamous cell carcinoma [Citation30,Citation34,Citation35] and cutaneous T-cell lymphoma [Citation36,Citation37]. Bacterial infections may compromise the anticancer therapy outcome due to inflammation in the tissue microenvironment and accelerated carcinogenesis [Citation38–42]. Hence, if an anticancer drug exhibits simultaneously antineoplastic and antibacterial effect, this would be beneficial for the oncological patients. In this regard, ERF keeps a significant capacity as a drug candidate for i.v. and topical clinical application in malignancies complicated by infections. The antimicrobial potential of ERF remains so far unexplored, although this will be especially important for its future clinical development.
The use of drug combinations for chemotherapy is a widely accepted approach to enhance drug efficiency and to reduce chemoresistance development upon antimicrobial and anticancer therapies. MLT and ERF are suitable candidates for drug combinations because they lack bone marrow toxicity and, depending on their critical micellar concentrations, they could include other drugs into the micellar structures. Appropriate drug combinations containing MLT could help enhance its moderate antimicrobial activity and achieve a bactericidal effect at lower concentrations. An attractive candidate for combination with MLT or ERF is the highly active and nontoxic natural product curcumin. Found in the root system of Curcuma longa (turmeric), curcumin (CRM) has been used for centuries in traditional medicine as anti-inflammatory, antineoplastic and antimicrobial agent [Citation43,Citation44]. However, several important pharmacokinetic problems hinder its successful clinical application: (i) insufficient absorption due to low water solubility, (ii) rapid intracellular metabolism and (iii) rapid systemic elimination [Citation45,Citation46]. These disadvantages reduce the bioavailability and compromise the expected therapeutic effect of CRM. One of the most successful possibilities to improve the aqueous solubility of CRM and to protect the molecule from rapid degradation is to encapsulate it in nano-delivery systems [Citation47–51]. Nano-sized CRM has already been successfully combined with MLT for treatment of leishmaniosis; thus giving the combination promising perspectives [Citation52].
Based on analysis of the published scientific data, we set the goal to explore the antibacterial activity of ERF against the Gram-positive bacterial species S. aureus, which is a common causative agent of skin infections being well known for its capacity to develop antimicrobial resistance [Citation53,Citation54]. S. aureus is a leading cause of biofilm-associated infections, which are usually long lasting and frequently occur in hospitals [Citation55]. As biofilm formation plays a crucial role for the survival of the bacterial population [Citation56], we investigated the biofilm inhibitory effects of MLT and ERF on methicillin resistant staphylococci, which have not been studied yet. Moreover, we aimed to improve the water solubility and to prolong the intracellular life of CRM by encapsulating it in nano-micelles based on Pluronic® P123 or a mixture of Pluronic® P123 and Pluronic® F127. In order to enhance the antibacterial activity of ERF or MLT and to highlight new challenging perspectives for their successful clinical use, we combined them with CRM loaded micelles based on strong theoretical evidence.
Materials and methods
Drugs and chemicals
The following chemicals and reagents were purchased from Sigma® Life Science, Steinheim, Germany: curcumin (#C1386, Mw = 368.385 g/mol), absolute ethanol (#46139), MLT (#M5571, Mw = 407.576 g/mol), Penicillin G potassium salt (#P3032, Mw = 356.37 g/mol), PBS (Dulbecco’s Phosphate Buffered Saline, #D8537) and 3–(4,5–dimethylthiazolyl-2)-2,5–diphenyltetrazolium bromide (#M2128, MTT dye). CRM (10 mmol/L) and Penicillin G (40 mg/L) were dissolved prior to use in absolute ethanol (CRM-Et) and in distilled water, respectively. A working solution of gentamycin (40 mg/L) was prepared from a ready-to-use injection solution (amp. 40 mg/mL, 1 mL, Sopharma®, Bulgaria). A 10 mmol/L stock solution of MLT was dissolved in ethanol/PBS (1:1, v/v) and stored at 4 °C. ERF (Mw = 503.74 g/mol), an APC synthesized by Prof. Hans-Jörg Eibl, MPI-Göttingen, Germany [Citation5], was stored at 4 °C as a 20 mmol/L stock solution in 0.9% NaCl.
Preparation of curcumin loaded micelles
The micelles were prepared by three methods: dissolution, solvent evaporation and film hydration method. The dissolution method was performed according to a previously described procedure [Citation57]. Briefly, the copolymers Pluronic® P123, Pluronic® F127 or their mixture at a 1:1 ratio were dissolved in purified water (10 mg/mL) and CRM was added to the solution under heating (37 °C). The dispersions were stirred at 700 rpm for 1 hour, filtered (0.22 µm) and kept at room temperature in a dark package.
The solvent evaporation method included dissolution of copolymers in purified water and addition of CRM dissolved in ethanol. After 1 h incubation, the ethanol was evaporated under vacuum and the aqueous micellar dispersions were filtered (0.22 µm) and kept for further studies.
The protocol of the film hydration method consisted of simultaneous dissolution of CRM and respective copolymer (or their mixture) in methanol. After complete evaporation of methanol, the films were dispersed in destilled water to give aqueous micellar dispersions (NCRM). The dispersions were filtered (0.22 µm) and kept for further studies.
Characterization of curcumin loaded micelles
The loading of CRM was calculated as a difference between its initial concentration and the concentration found in the fractions collected after rinsing of the filters used for filtration of the freshly loaded dispersions. CRM was determined by UV-Vis spectrophotometry at a wavelength of 428 nm (Thermo Fisher, USA). The standard curve was prepared in the concentration range of 2–10 µg/mL (r > 0.9994).
The size and zeta-potential were determined by photon correlation spectroscopy and electrophoretic laser Doppler velocimetry (Zetamaster analyzer, Malvern Instruments, UK). Freshly prepared micellar dispersions were measured at 25 °C with a scattering angle of 90°.
In vitro release of CRM from the three types of micelles was examined in a phosphate buffer (pH 7.0). The freshly prepared micellar dispersion was introduced into a dialysis membrane bag (MW = 6000–8000) that further was placed into 100 mL of PBS containing 10% ethanol. The release medium was stirred (50 rpm) and the temperature was maintained constant during the study (37 °C). At predetermined time intervals (2, 8 and 24 hours) samples were withdrawn from the medium outside the dialysis bag and the concentration of the released CRM was determined by UV-Vis spectrophotometry as described above.
Bacterial strains and growth conditions
Two strains S. aureus were investigated for susceptibility in the present study: ATCC 6538 (penicillin and methicillin sensitive, MSSA) and NBIMCC 8327 (methicillin resistant, MRSA). The strains were maintained in Trypticase Soy Agar/Broth (TSA/TSB, Himedia, India) at 37 °C, aerobic conditions. All experiments were performed in Mueller Hinton broth (MHB, #M0405B) or agar (MHA, #CM0337B), both from Thermo Scientific-Oxoid (UK).
Determination of minimal inhibitory and bactericidal concentrations
The minimal inhibitory concentrations (MIC) of CRM-Et, NCRM micelles and APCs were determined via the broth microdilution method (BMD) according to ISO 20776/1–2006 [Citation58]. Briefly, a bacterial suspension with density of 108 CFU/mL (OD600) was prepared from an overnight liquid bacterial culture and brought thereafter to a working bacterial suspension (WBS) with a concentration of 5 × 105 CFU/mL in MHB. Twofold serial dilutions were prepared in six-fold repetitions for each tested drug in 96 well plates to a volume of 50 µL. MHB was used as diluent and as test for absence of contamination. An equivalent volume of the WBS was added to each well to achieve final bacterial density of 5 × 104 CFU/mL. The plates were incubated 24 h at 37 °C. The lowest drug concentration, which prevented visible bacterial growth, was determined as MIC. Penicillin G (0.004–2 mg/L) and gentamycin (0.008–4 mg/L) were used as referent antibiotics (positive controls). The requirements of EUCAST (European Committee on Antimicrobial Susceptibility Testing) for their MICs were followed to discuss the results [Citation59]. PBS served as the negative control.
Samples from the BMD test treated with MIC and the remaining concentrations higher than MIC were sub-cultured in triplicate on MHA for 24 h at 37 °C (100 µL/Petri dish). The minimal bactericidal concentration (MBC) was defined as the lowest drug concentration reducing colony growth of the initial bacterial inoculum by ≥99.9%.
Cell redox activity test
The cell redox activity of all samples from the BMD assay was measured at the end of the incubation period with the MTT dye, which is reduced by the membrane located bacterial enzyme NADH:ubiquinone reductase (H+-translocation) to non-soluble violet crystals of formazan. The protocol of Wang et al. [Citation60] was applied after minor modifications for S. aureus. Briefly, 10 µL МТТ (5 mg/mL in PBS) was added to each sample, mixed thoroughly and incubated for 120 min at 37 °C. The resulting formazan was dissolved with an equivalent volume of 2-propranol containing 5% formic acid (HCOOH). The absorbance was measured at 550 nm (Absorbance Microplate Reader Lx800, Bio-Tek Instruments Inc., USA) against a blank solution containing respective volumes of MHB, MTT and solvent.
Mathematical modelling of redox activity after single drug treatment
The quantitative evaluation of the inhibition effects of the three drugs on both staphylococcal strains were performed by using two models considering an inhibitory effect of the applied concentration of drugs and fitting the experimental data.
The first model was adapted from the well-known Michaelis–Menten (MM) enzyme kinetics studies where an assumption of competitive inhibition as a mode of action (which is generally but not always true) was accepted. In our case the assumption is valid that the influence of drugs on microorganisms appeared on the population level. Hence, the model can be represented as follows
(1)
(1)
where Vm stands for overall (on the population level) normalized maximum enzyme activity, [%]; ‘Dose’ stands for concentration of drug, [µmol/L]; Ki is the inhibitory constant, which may be interpreted as IC50 in medical studies, [µmol/L];
The second model was particularly chosen because of its application to describing experimental data of similar studies. The Lambert–Pearson (LP) model is used to fit the experimental profile of the study of inhibition [Citation61]. In our case, the assumption is valid that the influence of drugs on microorganisms appeared on the population level. Hence, the model can be represented as follows
(2)
(2)
where Fa stands for normalized maximum enzyme activity, [%]; ‘Dose’ stands for concentration of drug, [µmol/L]; P1 is the inhibitory constant, which may be interpreted as IC50 in medical studies, [µmol/L]; and P2 stands for a slope.
The non-linear identification procedure was used to obtain the values of two model parameters: MM modified model (Vm and Ki) and LP model (P1 and P2) by fitting experimental data on the base of weighted least square statistical method (see and ). The program was coded in MAPLE symbolic mathematics software.
Biofilm formation assay
All drugs and CRM formulations were investigated for their potential to prevent biofilm formation of MRSA following the protocol of Stepanovic et al. [Citation62]. Twofold serial dilutions of the tested formulations were prepared in 96 well polystyrene tissue culture plates in BHI containing 2% glucose (w/v). The final volume was 100 µL/well. The drug concentrations ranged from 0 to 200 µmol/L. The bacterial inoculum was prepared the same way as described above for the BMD and an equivalent volume (100 µL) was added to each well. Cells were incubated at 37 °C for 24 h under static conditions. Thereafter, the supernatant was removed from each well and planktonic cells were removed by washing three times with PBS (250 µL/well). The remaining cells were fixed with methanol (200 µL for 15 min), air dried and stained with 0.1% crystal violet (200 µL/well). Excess stain was rinsed off with tap water and air dried. Biofilm formation was documented microscopically (40× magnification). The dye bound to biofilm was re-solubilized with 160 µL of 33% acetic acid and the optical density (OD) of each well was measured at 550 nm. The minimum biofilm inhibition concentration (MBIC50) was defined as the lowest concentration of the tested drugs that led to 50% inhibition on the biofilm formation. The biofilm inhibitory concentrations (BIC) were calculated in the program software GraphPad Prism with a mathematical model for dose–response relationship (variable slope) after normalization of the data and logarithmic transformation of the applied concentrations (X-data).
Checkerboard BMD test
The checkerboard BMD test was used for the in vitro evaluation of drug combinations, wherein two antimicrobial agents were serially diluted in a two-dimensional fashion to include all combinations within a specified clinically relevant range. Briefly, the BMD test was performed as described above. The BMD panels with the drug combinations were prepared using the following concentrations of CRM-Et, NCRM and APC (MLT or ERF) in doubling dilutions: from 250 μmol/L to 3.9 μmol/L. MICs were read after 24 h incubation. The results were interpreted following the fractional inhibitory concentration (FIC) methodology described below.
Fractional inhibitory concentration methodology
The FIC methodology was chosen for evaluation of drug–drug interactions because of several advantages: easy to be performed, very reliable; cost effective and accurate. It is based on determination of FICs by comparing the MIC of each drug alone to the MIC of that drug in combination with the second drug (MICC). Most commonly, synergy is usually defined as a four-fold decrease in the MIC of the drugs in combination when compared to the drugs tested alone. For calculation of FICs we used the MIC values obtained by the checkerboard BMD assay. The FICs were calculated and interpreted as follows [Citation63]:
Step 1
(3)
where FIC stands for fractional inhibitory concentration, A stands for drug A (MLT or erufosine) and B stands for drug B (CRM as ethanol solution or nano-particles).
Step 2
Synergy was defined as ƩFIC ≤ 0.5, indifference as 0.5<ƩFIC ≤ 4 and antagonism as ƩFIC > 4. Some investigators [Citation63] consider compounds additive when 0.5<ƩFIC ≤ 1, which was adopted in this study.
Statistics
The program software GraphPad Prism (Version 6.0 for Windows, GraphPad Software, La Jolla California, USA) was used for statistical analysis of the experimental data. Statistical differences and significance were assessed by the Student’s t-test. Each experiment was performed in triplicate. Minimum three samples for each concentration, the positive, negative and untreated controls were analyzed.
Results and discussion
In the present study, we report for the first time the antibacterial activity of ERF against MSSA and MRSA strains and the biofilm inhibitory potential of ERF and MLT, against MRSA. We managed to enhance the moderate anti-staphylococcal activity of both APCs through combinations with the natural product CRM in a suitable ratio. In order to improve the water solubility of CRM and to overcome the problems related to its poor bioavailability and fast metabolism, the molecule was encapsulated in micellar nano-delivery systems based on Pluronic® 123 and Pluronic® 127.
Physicochemical properties of the micelles
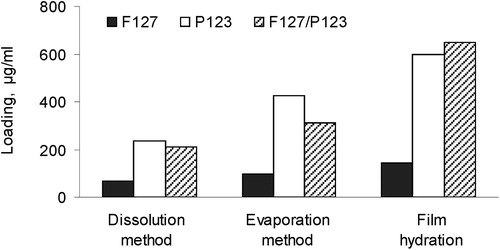
CRM loading in Pluronic® micelles was performed by three methods, namely, in particular dissolution, solvent evaporation and film hydrating method. The developed micelles were based on Pluronic® P123, Pluronic® F127 or their combination (P123/F127). represents the influence of both the method and the properties of the respective Pluronic® type on the loading of CRM into the micelles. The lowest loading degree was observed in micelles based on Pluronic® F127, which was explained with the more hydrophilic properties of this Pluronic® type. Higher loading was achieved in Pluronic® P123 and mixed micelles, which was probably due to the high affinity between the more hydrophobic Pluronic® P123 and CRM. Regarding the influence of the method, the film hydration method resulted in a higher loading degree of CRM in all types of the micelles. In particular, the loading achieved by the film hydration method was 2.5 and 3 times higher (for P123 and mixed micelles, respectively) than the loading achieved by the dissolution method. Even in the case of the more hydrophilic F127 carrier, the loading was two times higher than with the dissolution method. Thus, the film hydration method was considered the most appropriate method for CRM loading into the selected carriers.
Figure 1. Loading of curcumin (µg/mL micellar dispersion) depending on the loading method and Pluronic® type.
The physicochemical properties of all types of CRM loaded micelles (NCRM) are shown in . The micelles formulated with Pluronic® P123 had smaller diameter and narrow size distribution. The micelles based on Pluronic® F127 and the mixture with participation of Pluronic® F127 showed a tendency for augmented size. However, the mean diameter of all micelles did not exceed 100 nm, which was considered advantageous for their stability and in vivo performance. Regarding zeta-potential, all types of micelles possessed slightly negative values for zeta-potential, which correlated with the known non-ionic properties of Pluronic® copolymers.
Table 1. Size and zeta-potential of the developed micelles loaded with CRM.
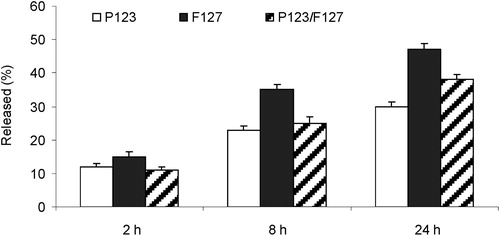
The in vitro release study was performed in a phosphate buffer (pH = 7.0). The release of CRM was faster from Pluronic® F127 micelles than from Pluronic® P123 and mixed micelles (). The slower release rate from these micelles corresponded to the higher affinity of CRM to Pluronic® P123 type, which retained CRM in the hydrophobic core of both types of micelles containing Pluronic® P123.
Figure 2. In vitro release of curcumin in a phosphate buffer.
Because of the observed better characteristics of the micelles based on Pluronic® P123, further experiments were performed with Pluronic® P123 (NCRM1) and mixed P123/F127 (NCRM2) micelles. The experiments were related to evaluation of antibacterial activity of micellar CRM.
MIC and MBC after single drug treatment
The data from the BMD test suggested that ERF achieves MIC against the two tested staphylococcal strains at clinically relevant [Citation33] concentrations (). The MIC of ERF on methicillin resistant staphylococci was by 10 µmol/L higher (60 µmol/L) than that measured in the MSSA strain ATCC 3865 (50 µmol/L). Nevertheless, the colony growth inhibition at MIC in MRSA (, ) was remarkably stronger (only 10 surviving CFU/mL) than that obtained for the sensitive staphylococci (1.7 × 103 CFU/mL). The susceptibility of the MSSA strain to MLT was higher in our study than that reported by Lukac et al. [Citation64] in 2012 for the same strain. ERF inhibited the colony growth of MSSA and MRSA to 10 CFU/mL at lower concentrations (62.5 and 60 µmol/L, respectively) than MLT (100 µmol/L) as presented in . Regarding the MBC, both APCs achieved bactericidal effect at very high concentrations, which are not clinically acceptable: 500 µmol/L for the MRSA strain and 500 µmol/L (MLT) or 1000 µmol/L (ERF) for ATCC 3865. Based on this result, we can assume that both drugs exhibit a bacteriostatic effect. In line with the findings of Obando et al. [Citation16], ERF showed a stronger effect on MRSA than MSSA regarding the number of CFU/mL and the MBC values. As evident from the lower MICs and the number of surviving bacteria at MIC for both test-strains (), ERF showed a better colony growth inhibitory effect than MLT in concentrations lower than 500 µmol/L. In this case, the result obtained for the antibacterial effect of erufosine does not support the general observation of Lukac et al. [Citation64] that APCs forming micellar aqueous solutions (MLT) exhibit stronger antimicrobial activity than APCs forming lamellar solutions in water (ERF). However, we cannot draw a definitive conclusion that ERF is more potent in its antibacterial activity than MLT before testing on other bacterial strains.
Figure 3. Effect of single drugs on colony formation.
Note: CRM-Et, ethanol solution of curcumin; MLT, miltefosine; ERF, erufosine.
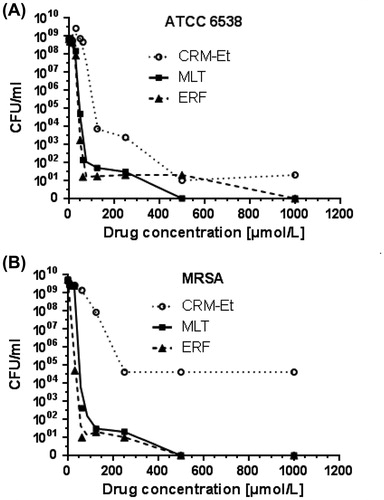
Table 2. Antibacterial activity of examined drugs against MSSA and MRSA.
CRM was found to be effective against S. aureus in numerous investigations. The MIC value of non-encapsulated CRM measured in our study for ATCC 6538 (125 µmol/L, 46 mg/L) falls within the general range reported for other MSSA strains (18.42–256 mg/L) treated with ethanol or DMSO solutions of the compound [Citation65,Citation66]. Hence, the MIC estimated by the BMD method in our study was significantly (three to fourfold) lower than the values (125–217 mg/L) reported by other authors for ethanol solutions and was comparable to the MIC values of DMSO solutions [Citation65]. This variation could be due to the specific sensitivity of different staphylococcal strains. Interestingly, in our experiments, we did not observe any difference in the MIC values resulting from the accessibility of the MSSA and MRSA strains to free CRM. The growth of MRSA was inhibited at the same concentration as that of MSSA (). Regarding the number of CFU/mL at MIC, the MSSA strain was negligibly more sensitive (7.2 × 103 CFU/mL) than the MRSA strain with its ∼105 CFU/mL (, ).
Recently, Mun et al. [Citation67] showed that treatment of MRSA with CRM in the presence of membrane permeability enhancers and ATPase inhibitors leads to damage of the cell wall, disruption of the cytoplasmic contents, and cell lysis. The authors proved by western blotting that CRM significantly lowers the protein level of PBP2a (penicillin binding protein 2a) in MRSA, which could be a possible explanation for the equal sensitivity of the MSSA and MRSA strain in our study. In general, CRM was less effective on both test-strains than ERF and MLT. As presented in , the compound exhibited bacteriostatic effect. The maximum inhibitory effect for MSSA was 10 CFU/mL at a concentration of 500 µmol/L versus 5 × 104 CFU/mL for MRSA achieved at 250 µmol/L and remained unchanged irrespective of the increase in concentration.
The antibacterial effect of both NCRM-micelles produced and tested within the present study was equal for both staphylococcal test-strains, but significantly lower (MIC = 500 µmol/L, 184 mg/L) than for free CRM. This result is logical and expected, considering the release time of CRM from the micelles. For a period of 24 hours, which coincides with the time of treatment of the pathogens, the P123 micelles released approximately up to 30% (150 µmol/L, 55.2 mg/L), whereas the mixed micelles released up to 38% (190 µmol/L, 69.9 mg/L) of the loaded CRM (). The fact that the MICs of NCRM remained still slightly higher than those of the ethanol formulation, leads to the suggestion that the effect of CRM was concentration-dependent rather than time-dependent.
Inhibition of the bacterial redox activity after single drug treatment
CRM, MLT and ERF exhibited similar potential to inhibit the respiratory metabolism of both test-strains (, and ). Only for CRM, there was a slight difference between the redox activity of the MRSA strain (13.8% redox activity) compared to the MSSA one (9.2% redox activity), which did not have an impact on the MIC of the compound but resulted in a stronger inhibition of colony formation.
Figure 4. Redox activity of S. aureus strains after single drug treatment – MM modified model.Note: NEA, normalized enzyme activity, [%]; Vm, overall normalized maximum enzyme activity, [%]; Ki, inhibition constant, [µmol/L]; R, correlation coefficient, [-].
![Figure 4. Redox activity of S. aureus strains after single drug treatment – MM modified model.Note: NEA, normalized enzyme activity, [%]; Vm, overall normalized maximum enzyme activity, [%]; Ki, inhibition constant, [µmol/L]; R, correlation coefficient, [-].](/cms/asset/af94cf7e-7efc-4643-9215-344e9ed290a4/tbeq_a_1533792_f0004_c.jpg)
Figure 5. Redox activity of S. aureus strains after single drug treatment – LP model.
Note: NEA, normalized enzyme activity; [%]; Dose, concentration of drug, [µmol/L]; P1, inhibitory constant, which may be interpreted IC50 in medical studies or as the dose achieving 50% inhibition, [µmol/L]; P2, slope, [-]; R, correlation coefficient, [-].
![Figure 5. Redox activity of S. aureus strains after single drug treatment – LP model.Note: NEA, normalized enzyme activity; [%]; Dose, concentration of drug, [µmol/L]; P1, inhibitory constant, which may be interpreted IC50 in medical studies or as the dose achieving 50% inhibition, [µmol/L]; P2, slope, [-]; R, correlation coefficient, [-].](/cms/asset/8ef5ee65-c3ec-458b-9b26-399342bf8dfc/tbeq_a_1533792_f0005_c.jpg)
Two mathematical models for calculating the redox inhibitory potential of CRM, ERF and MLT were compared. The MM modified model simulation profiles in MAPLE software and experimental data are presented in . Analyzing the results from , we felt confident that the accepted hypothesis of inhibition and the adapted model gave excellent results according to the correlation coefficient R given below each graph. It is noteworthy that the best fitting with the MM model gave R in the range of 0.94–0.95 () for S. aureus ATCC 6538 and 0.93–0.96 () for the MRSA strain, using three single drugs.
Analysing the simulation results of the LP model and the same experimental data in , one can see that this model fitted them much better (A, B). The LP model is successfully used to describe many bacterial processes where inhibition on bacterial population takes place. Hence, the R coefficient values obtained with the LP model showed a quasi-perfect match between the concentration profiles and the experimental ones and R was in the range of 0.995–0.998 () for S. aureus ATCC 6538 and 0.997–0.998 () for the MRSA strain, using three single drugs.
Neither of the two NCRM formulations showed redox inhibitory effects as strong as CRM (); thus, confirming the result from the BMD tests. The metabolic activity of the treated bacteria ranged from 51.2 (NCRM1) to 56% (NCRM2) for MSSA and from 61 (NCRM2) to 63.5% (NCRM1) for MRSA. Evidently, the observed difference between both NCRM formulations was negligible, but significantly higher as compared to the samples treated with CRM-Et (9.2% for MSSA and 13.8% for MRSA). This could be due to the gradual release of smaller doses of CRM from the micelles, which possibly are not sufficient to suppress the metabolism of the bacterial cells as efficiently as the single administration of the whole dose.
Micellar curcumin, miltefosine and erufosine inhibit the biofilm formation of MRSA
Biofilm formation represents the most important microbial defence strategy for survival of a bacterial population. The key role of biofilms in antibiotic resistance and failure of the immune system response is increasingly recognized in the recent decade as a significant risk to public health [Citation55]. Biofilms are involved in over 80% of the microbial infections in the body and can cause significant clinical problems, especially in immunocompromised patients, by impairing the antibiotic treatment and resulting in chronic disease [Citation68]. Most of the classical antibiotics are designed to destroy fast-growing metabolically active bacteria in aerobic conditions, whereas biofilms consist of multiple layers of slow growing antibiotic tolerant microorganisms in a low metabolic state [Citation56]. The pathogenic bacterial species S. aureus appears to possess a strong adhesion function and is well-known for its capability to form biofilms by attaching to substrate surfaces [Citation69–71]. Current strategies to combat this medical problem include killing of the bacteria, mechanical removal of the biofilm or chemical degradation by chelating complexes and surfactants. Nevertheless, the selection of a successful treatment is still a significant challenge [Citation55].
In the present study, we demonstrated for first time that MLT and ERF exert a strong inhibitory effect on the formation of MRSA biofilm at low (5–30 µmol/L), clinically relevant concentrations (). The MBIC50 of ERF was threefold lower (1.87 µmol/L) than that of MLT (6 µmol/L), as presented in and . The morphological evaluation of the MRSA biofilm () demonstrated that MLT destroyed the biofilm layer to a greater extent than ERF at concentrations of 10 µmol/L, but ERF continued to inhibit the biofilm formation significantly at lower concentrations (5 µmol/L) unlike MLT. At concentrations higher than 10 µmol/L both APCs exerted the same effect, achieving full biofilm inhibition at 20 µmol/L. As known from the published literature [Citation71], the S. aureus biofilm development proceeds in three steps: initial adhesion, proliferation and detachment, detailed exploration of which regarding the activity of MLT and ERF was not the subject of this study. In order to clarify the complex signal transduction underlying the biofilm inhibitory effect of both APCs, further in-depth investigations should be performed in a separate study. Nevertheless, the potential of these drugs to destroy MRSA biofilm, as revealed in the present study, provides useful information relevant to their clinical administration and of important benefit in the treatment of skin malignancies accompanied by chronic wound/lesion infections.
Figure 6. Quantitative biofilm evaluation after treatment with ethanol solution of curcumin, curcumin-loaded micelles, miltefosine and erufosine.
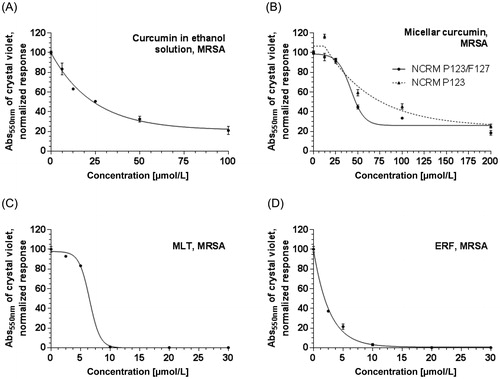
Figure 7. Biofilm formation of MRSA after treatment with curcumin (in the form of ethanol solution or nano-micelles) and alkylphosphocholines: microscopic documentation of the biofilm inhibition.
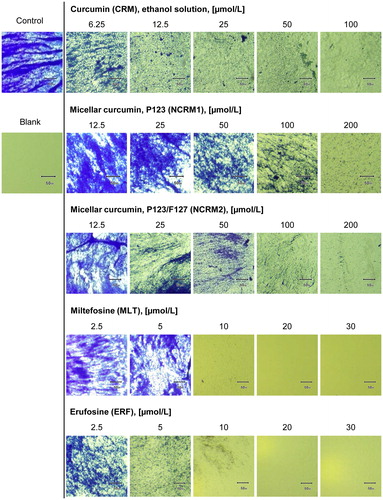
The results for the formulations NCRM1 and NCRM2 (, and ) revealed that micellar CRM could break down up to 90% of the MRSA biofilm at 200 µmol/L (73.7 mg/L), which corresponds to 30% (60 µmol/L, 22.1 mg/L) and 38% (76 µmol/L, 28 mg/L) CRM released from the P123 and P123/F127 micelles, respectively. In comparison, free CRM removed the biofilm to a lesser extent (up to 80%) at the highest tested concentration, 100 µmol/L (36.8 mg/L), but achieved a lower MBIC50 (24.84 µmol/L, 8.83 mg/L). Hence, the effect of NCRM on biofilm formation regarding the highest tested concentrations was stronger than that of pure CRM, in contrast to the MIC, MBC, CFU/mL, redox activity and MBIC measured in the present study. At concentrations lower than 100 µmol/L, CRM had stronger biofilm inhibitory effect than NCRM. A possible explanation for this discrepancy is that the polymers, which possess properties similar to those of the surfactants, contributed to the detachment of the bacteria from the experimental surface and in this way increased the effect of CRM in reducing the biofilm layer. It is noteworthy that micellar CRM based on P123/F127 was a stronger biofilm inhibitor (MBIC = 62 µmol/L, 22 mg/L) than the CRM-loaded micelles based on P123 only (MBIC = 88.2 µmol/L, 32.5 mg/L).
Micellar curcumin potentiates the antistaphylococcal activity of APCs
Considering the moderate antibacterial effect of MLT and ERF, we aimed at improving their activity through combinations with the non-toxic compound CRM at different ratios applied in the BMD checkerboard assay. The checkerboard BMD is the most frequently used method and a milestone in the synergy strategy because it is a less labour-intensive approach to assess antibiotic interactions in vitro than the time-kill assay. Changing the concentrations of all drugs within the combinations, it was possible to find the drug ratios that led to synergistic interactions in both test strains. shows the combinations with synergistic, additive and indifferent effects. All other combinations, which led to antagonistic interactions, are not shown. Combinations between MLT and NCRM in MRSA managed to lower the MICC of MLT by twofold, and the MICC of NCRM by 16-fold, thus achieving synergism at a ratio of 1:1 versus the MIC values of the single drugs. Similar results were observed only for the combination of ERF with NCRM2, but not with NCRM1. It is noteworthy that increasing the concentration of CRM-Et in the MLT:CRM combination from 16:1 to 1:1, without changing the concentration of MLT, did not lead to synergy, unlike the combination with NCRM. Moreover, the MICC of MLT remained unchanged as compared to the MIC value obtained after single drug application. The MICC of CRM in this interaction decreased 32 times versus the MIC in response to CRM monotherapy. The same trend was observed for ERF, except that only one combination (ERF:NCRM2 in a ratio of 1:1) led to an additive interaction and to a twofold decrease of the MICC observed after ERF alone. For the other two combinations (ERF:CRM and ERF:NCRM1), the measured effect was indifference (), wherein the MICC of ERF remained unchanged versus the MIC of the single drug. Evidently, the combinations with CRM-Et or NCRM1 did not diminish the MIC of ERF.
Table 3. Antibacterial activity of drug combinations.
The MSSA strain was not as sensitive to the tested combinations as the MRSA strain. Lower MICC values of APCs were related to additive effects, but not synergism. Additive interactions were observed only in the combinations with NCRM formulations ()—both micellar systems combined with ERF or MLT with NCRM1.
The analysis of the drug–drug interactions in revealed that synergistic or additive effects occurred only if there was a decrease in the MICC of the APCs. In all combinations, CRM and NCRM were characterized by significantly lower MICC as compared to the single application, and changes in their values obviously did not influence the final effect of the combination. Synergism was achieved only after using NCRM formulations, but not CRM-Et.
In parallel to both APCs, we also included the antibiotic penicillin in the combination experiments as a reference combination for the MSSA strain. Additive interaction and synergy were observed when combining penicillin with NCRM1 and NCRM2, respectively. On the contrary, the combination with free CRM increased the MICC of penicillin versus the MIC value of the single antibiotic resulting in an indifferent effect. Based on the published data on the mode of action of CRM [Citation67] and penicillin [Citation72], we could assume that this result was related to the interaction of both compounds with the penicillin binding proteins (PBPs). These transpeptidases are important targets of penicillin, which blocks the cell wall synthesis after binding to them [Citation72]. According to Mun et al. CRM suppresses the mecA gene encoding the PBP2a protein with low penicillin affinity and as a result, MRSA can be sensitized towards the antibacterial action of β-lactam antibiotics such as penicillin and methicillin [Citation65,Citation73]. Recently, it was found that CRM also binds to the PBPs in Streptococcus pneumoniae [Citation74]. Based on these data, it can be assumed that CRM and penicillin compete for interaction with the same target proteins, which impedes the achievement of an additive or synergistic effect. The successful potentiation of the penicillin effect in the presence of NCRM could be explained by the prolonged release of smaller CRM doses from the nano-micelles. In this way more PBP-molecules remain free and available for interaction with the antibiotic, thereby leading to a stronger combination effect.
Conclusions
The estimated anti-staphylococcal and biofilm inhibitory activities of ERF and MLT widen the spectrum of their useful pharmacological effects. Both APCs showed remarkable ability to remove MRSA biofilm at lower clinically relevant concentrations, which could be of important benefit for immunocompromised cancer patients with impaired barrier function of the skin and infected skin wounds/lesions. CRM encapsulated in P123/F127 micelles improved significantly the moderate antibacterial effect of MLT and ERF in MRSA at a ratio of 1:1; thus rising new possibilities for the development of effective drug combinations. The LP kinetic model coded in MAPLE® for quantitative evaluation of the redox activity data showed a quasi-perfect match with the experimental points, which demonstrated its suitability for such analyses. MLT and ERF strongly suppressed the respiratory metabolism of MRSA resulting in less than 103 CFU/mL. Finally, the simultaneous antineoplastic and antimicrobial potential and the established synergistic and additive drug combinations reveal new horizons in the application of ERF and MLT as antineoplastic agents for the treatment of numerous malignancies with skin lesions, which are accompanied by chronic bacterial infections.
Acknowledgements
The authors thank Dr. Stanislav Phillipov, Ph.D. (Chair Human Anatomy, Histology, General and Clinical Pathology and Forensic Medicine, Faculty of Medicine, University Hospital Lozenetz, Sofia University “St. Kliment Ohridski”, 2 Kozyak Str, Sofia, Bulgaria) for his help in performing the microscopic pictures.
Disclosure statement
The authors declare that they have no competing interests.
Funding
This study was supported by the Bulgarian National Science Foundation under grant number DN 03/3 (2016–2018). The redox activity and checkerboard tests were performed using laboratory equipment donated by the Alexander von Humboldt Foundation (Grant “Equipment Subsidies“) to Maya M. Zaharieva, Ph.D., for establishing a “Laboratory for in vitro cytotoxicity and signal transduction” at the Stephan Angeloff Institute of Microbiology, Bulgarian Academy of Sciences.
Related Research Data
References
- Dorlo TP, Balasegaram M, Beijnen JH, et al. Miltefosine: a review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J Antimicrob Chemother. 2012;67:2576–2597.
- Jaffrés P-A, Gajate C, Bouchet AM, et al. Alkyl ether lipids, ion channels and lipid raft reorganization in cancer therapy. Pharmacol Ther. 2016;165:114–131.
- Rios-Marco P, Marco C, Galvez X, et al. Alkylphospholipids: an update on molecular mechanisms and clinical relevance. Biochim Biophys Acta Biomembr. 2017;1859:1657–1667.
- Berger MR, Yanapirut P, Reinhardt M, et al. Antitumor activity of alkylphosphocholines and analogues in methylnitrosourea-induced rat mammary carcinomas. Prog Exp Tumor Res. 1992;34:98–115.
- Eibl H, Hilgard C, Unger C, editors. Alkylphosphocholines: New drugs in cancer therapy. Basel (Switzerland): Karger; 1992. (Prog Tumor Res, Vol. 34).
- Murray HW, Berman JD, Davies CR, et al. Advances in leishmaniasis. Lancet. 2005;366:1561–1577.
- Varela MR, Villa-Pulgarin JA, Yepes E, et al. In vitro and in vivo efficacy of ether lipid edelfosine against Leishmania spp. and SbV-resistant parasites. PLoS Negl Trop Dis. 2012;6:e1612. DOI: 10.1371/journal.pntd.0001612
- Le Fichoux Y, Rousseau D, Ferrua B, et al. Short- and long-term efficacy of hexadecylphosphocholine against established Leishmania infantum infection in BALB/c mice. Antimicrob Agents Chemother. 1998;42:654–658.
- Kuhlencord A, Maniera T, Eibl H, et al. Hexadecylphosphocholine: oral treatment of visceral leishmaniasis in mice. Antimicrob Agents Chemother. 1992;36:1630–1634.
- Coelho AC, Trinconi CT, Costa CH, et al. In vitro and in vivo miltefosine susceptibility of a Leishmania amazonensis isolate from a patient with diffuse cutaneous leishmaniasis. PLoS Negl Trop Dis. 2014 ;8:e2999. DOI: 10.1371/journal.pntd.0002999
- Konstantinov SM, Kaminsky R, Brun R, et al. Efficacy of anticancer alkylphosphocholines in Trypanosoma brucei subspecies. Acta Trop. 1997;64:145–154.
- Seifert K, Duchene M, Wernsdorfer WH, et al. Effects of miltefosine and other alkylphosphocholines on human intestinal parasite Entamoeba histolytica. Antimicrob Agents Chemother. 2001;45:1505–1510.
- Eissa MM, El-Azzouni MZ, Amer EI, et al. Miltefosine, a promising novel agent for schistosomiasis mansoni. Int J Parasitol. 2011;41:235–242.
- Choubey V, Maity P, Guha M, et al. Inhibition of Plasmodium falciparum choline kinase by hexadecyltrimethylammonium bromide: a possible antimalarial mechanism. Antimicrob Agents Chemother. 2007;51:696–706.
- Llull D, Rivas L, Garcia E. In vitro bactericidal activity of the antiprotozoal drug miltefosine against Streptococcus pneumoniae and other pathogenic streptococci. Antimicrob Agents Chemother. 2007;51:1844–1848.
- Obando D, Widmer F, Wright LC, et al. Synthesis, antifungal and antimicrobial activity of alkylphospholipids. Bioorg Med Chem. 2007;15:5158–5165.
- Widmer F, Wright LC, Obando D, et al. Hexadecylphosphocholine (miltefosine) has broad-spectrum fungicidal activity and is efficacious in a mouse model of cryptococcosis. Antimicrob Agents Chemother. 2006;50:414–421.
- Tong Z, Widmer F, Sorrell TC, et al. In vitro activities of miltefosine and two novel antifungal biscationic salts against a panel of 77 dermatophytes. Antimicrob Agents Chemother. 2007;51:2219–2222.
- Berger MR, Betsch B, Gebelein M, et al. Hexadecylphosphocholine differs from conventional cytostatic agents. J Cancer Res Clin Oncol. 1993;119:541–548.
- Berger MR, Muschiol C, Schmähl D, et al. New cytostatics with experimentally different toxic profiles. Cancer Treat Rev. 1987;14:307–317.
- Croft SL, Engel J. Miltefosine-discovery of the antileishmanial activity of phospholipid derivatives. Trans R Soc Trop Med Hyg. 2006 ;100 Suppl 1:S4–S8. DOI: S0035–9203(06)00197–0.
- Tsushima S, Yoshioka Y, Tanida S, et al. Syntheses and antimicrobial activities of alkyl lysophospholipids. Chem Pharm Bull. 1982;30:3260–3270.
- Achterberg V, Gercken G. Cytotoxicity of ester and ether lysophospholipids on Leishmania donovani promastigotes. Mol Biochem Parasitol. 1987;23:117–122.
- Croft SL, Neal RA, Pendergast W, et al. The activity of alkyl phosphorylcholines and related derivatives against Leishmania donovani. Biochem Pharmacol. 1987;36:2633–2636.
- German Drug Registration Authorities [Internet]. Impavido 10/50 mg Kapseln - Fachinformation. 2008 – [Cited 2018 Feb 1]. Available from: http://www.pharmnet-bund.de/dynamic/de/index.html
- Unger C, Sindermann H, Peukert M, et al. Hexadecylphosphocholine in the topical treatment of skin metastases in breast cancer patients. Prog Exp Tumor Res. 1992;34:153–159.
- Who Model List of Essential Medicines - 17th List. Geneva (Switzerland): World Health Organization; c2011 [Cited 2018 Feb 1]. Available from: http://www.who.int/medicines/publications/essentialmedicines/en/index.html
- Huelves L, Del Prado G, Gracia M, et al. In vitro and in vivo activity of miltefosine against penicillin-sensitive and -resistant Streptococcus pneumoniae strains. J Chemother. 2008;20:441–444.
- Konigs SK, Pallasch CP, Lindner LH, et al. Erufosine, a novel alkylphosphocholine, induces apoptosis in CLL through a caspase-dependent pathway. Leuk Res. 2010;34:1064–1069.
- Kapoor V, Zaharieva MM, Das SN, et al. Erufosine simultaneously induces apoptosis and autophagy by modulating the Akt-mTOR signaling pathway in oral squamous cell carcinoma. Cancer Lett. 2012;319:39–48.
- Fiegl M, Lindner LH, Juergens M, et al. Erufosine, a novel alkylphosphocholine, in acute myeloid leukemia: single activity and combination with other antileukemic drugs. Cancer Chemother Pharmacol. 2008;62:321–329.
- Martelli AM, Papa V, Tazzari PL, et al. Erucylphosphohomocholine, the first intravenously applicable alkylphosphocholine, is cytotoxic to acute myelogenous leukemia cells through JNK- and PP2A-dependent mechanisms. Leukemia. 2010;24:687–698.
- Yosifov DY, Todorov PT, Zaharieva MM, et al. Erucylphospho-N,N,N-trimethylpropylammonium (erufosine) is a potential antimyeloma drug devoid of myelotoxicity. Cancer Chemother Pharmacol. 2011;67:13–25.
- Ansari SS, Sharma AK, Zepp M, et al. Upregulation of cell cycle genes in head and neck cancer patients may be antagonized by erufosine's down regulation of cell cycle processes in OSCC cells. Oncotarget. 2017;9:5797–5810.
- Kapoor V, Zaharieva MM, Berger MR. Erufosine induces autophagy and apoptosis in oral squamous cell carcinoma: role of the Akt-mTOR signaling pathway. In: Hayat MA, editor. Autophagy: Cancer, other pathologies, inflammation, immunity, infection, and aging. Mitophagy. Vol.3, 1st ed. Amsterdam: Academic Press - Elsevier; 2014. p. 229–245.
- Yosifov DY, Kaloyanov KA, Guenova ML, et al. Alkylphosphocholines and curcumin induce programmed cell death in cutaneous T-cell lymphoma cell lines. Leuk Res. 2014;38:49–56.
- Zaharieva MM, Trochopoulos A, Dimitrov L, et al. New insights in routine procedure for mathematical evaluation of in vitro cytotoxicity data from cancer cell lines. Int J Bioautomation. 2018;22:87–106.
- Axelrod PI, Lorber B, Vonderheid EC. Infections complicating mycosis fungoides and Sézary syndrome. JAMA. 1992;267:1354–1358.
- de Martel C, Franceschi S. Infections and cancer: established associations and new hypotheses. Crit Rev Oncol Hematol. 2009;70:183–194.
- Moore MM, Chua W, Charles KA, et al. Inflammation and cancer: causes and consequences. Clin Pharmacol Ther. 2010;87:504–508.
- Willerslev-Olsen A, Krejsgaard T, Lindahl LM, et al. Bacterial toxins fuel disease progression in cutaneous T-cell lymphoma. Toxins (Basel). 2013;5:1402–1421.
- Zhu C, Wang Y, Cai C, et al. Bacterial infection and associated cancers. Adv Exp Med Biol. 2017;1018:181–191.
- Oliveira AS, Sousa E, Vasconcelos MH, et al. Curcumin: a natural lead for potential new drug candidates. Curr Med Chem. 2015;22:4196–4232.
- Perrone D, Ardito F, Giannatempo G, et al. Biological and therapeutic activities, and anticancer properties of curcumin. Exp Ther Med. 2015;10:1615–1623.
- Anand P, Kunnumakkara AB, Newman RA, et al. Bioavailability of curcumin: problems and promises. Mol Pharm. 2007;4:807–818.
- Sharma RA, Steward WP, Gescher AJ. Pharmacokinetics and pharmacodynamics of curcumin. Adv Exp Med Biol. 2007;595:453–470.
- Bhawana BRK, BHS, et al. Curcumin nanoparticles: preparation, characterization, and antimicrobial study. J Agric Food Chem. 2011;59:2056–2061.
- Ghalandarlaki N, Alizadeh AM, Ashkani-Esfahani S. Nanotechnology-applied curcumin for different diseases therapy. Biomed Res Int. 2014;2014:1–23. DOI: 10.1155/2014/394264
- Krausz AE, Adler BL, Cabral V, et al. Curcumin-encapsulated nanoparticles as innovative antimicrobial and wound healing agent. Nanomedicine. 2015;11:195–206.
- Gopal J, Muthu M, Chun SC. Water soluble nanocurcumin extracted from turmeric challenging the microflora from human oral cavity. Food Chem. 2016;211:903–909.
- Jahanizadeh S, Yazdian F, Marjani A, et al. Curcumin-loaded chitosan/carboxymethyl starch/montmorillonite bio-nanocomposite for reduction of dental bacterial biofilm formation. Int J Biol Macromol. 2017;105:757–763.
- Tiwari B, Pahuja R, Kumar P, et al. Nanotized curcumin and miltefosine, a potential combination for treatment of experimental visceral leishmaniasis. Antimicrob Agents Chemother. 2017;61:e01169–e01116. DOI: 10.1128/aac.01169–16
- Goncalves da Silva A, Baines SL, Carter GP, et al. A phylogenomic framework for assessing the global emergence and evolution of clonal complex 398 methicillin-resistant Staphylococcus aureus. Microb Genom. 2017;3:e000105. DOI: 10.1099/mgen.0.000105
- Monaco M, Pimentel de Araujo F, Cruciani M, et al. Worldwide Epidemiology and Antibiotic Resistance of Staphylococcus aureus. In: Bagnoli F, Rappuoli R, Grandi G, editors. Staphylococcus aureus: microbiology, pathology, immunology, therapy and prophylaxis. Cham: Springer; 2016. p. 21–56.
- Metcalf D, Bowler P, Parsons D. Wound biofilm and therapeutic strategies. In: Dhanasekaran D, editor. Microbial biofilms. London (UK): IntechOpen; 2016. DOI: 10.5772/63238
- Sonderholm M, Bjarnsholt T, Alhede M, et al. The consequences of being in an infectious biofilm: Microenvironmental conditions governing antibiotic tolerance. IJMS. 2017;18:2688. DOI: 10.3390/ijms.18122688
- Kabanov AV, Batrakova EV, Melik-Nubarov NS, et al. A new class of drug carriers: micelles of poly(oxyethylene)-poly(oxypropylene) block copolymers as microcontainers for drug targeting from blood in brain. J Controlled Release. 1992;22:141–157.
- ISO20776/1 –2006. Clinical laboratory testing and in vitro diagnostic test systems — Susceptibility testing of infectious agents and evaluation of performance of antimicrobial susceptibility test devices — Part 1: Reference method for testing the in vitro activity of antimicrobial agents against rapidly growing aerobic bacteria involved in infectious diseases. European Committee for Standardization (CEN), Technical Committee CEN/TC 140, Technical Committee ISO/TC 212. 2006. p. 19.
- EUCAST: managing infections promoting science [Internet]. Basel (Switzerland): The European Committee on Antimicrobial Susceptibility Testing - EUCAST; c2018. Clinical breakpoints. Breakpoint tables for bacteria; 2013 Apr 07 [cited 2018 Jun 23]. Available from: http://www.eucast.org/clinical_breakpoints/
- Wang H, Cheng H, Wang F, et al. An improved 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) reduction assay for evaluating the viability of Escherichia coli cells. J Microbiol Methods. 2010;82:330–333.
- Lambert RJW, Lambert R. A model for the efficacy of combined inhibitors. J Appl Microbiol. 2003;95:734–743.
- Stepanovic S, Vukovic D, Hola V, et al. Quantification of biofilm in microtiter plates: overview of testing conditions and practical recommendations for assessment of biofilm production by staphylococci. APMIS. 2007;115:891–899.
- Jenkins SG, Schuetz AN. Current concepts in laboratory testing to guide antimicrobial therapy. Mayo Clin Proc. 2012;87:290–308.
- Lukac M, Garajova M, Mrva M, et al. Relationship between aggregation properties and antimicrobial activities of alkylphosphocholines with branched alkyl chains. Int J Pharm. 2012;423:247–256.
- Teow SY, Liew K, Ali SA, et al. Antibacterial action of curcumin against Staphylococcus aureus: a brief review. J Trop Med. 2016;2016:1–10. DOI: 10.1155/2016/2853045
- Moghadamtousi SZ, Kadir HA, Hassandarvish P, et al. A review on antibacterial, antiviral, and antifungal activity of curcumin. Biomed Res Int. 2014;2014:1–12. DOI: 10.1155/2014/186864
- Mun SH, Kim SB, Kong R, et al. Curcumin reverse methicillin resistance in Staphylococcus aureus. Molecules. 2014;19:18283–18295.
- Davies D. Understanding biofilm resistance to antibacterial agents. Nat Rev Drug Discov. 2003;2:114–122.
- Makino T, Jimi S, Oyama T, et al. Infection mechanism of biofilm-forming Staphylococcus aureus on indwelling foreign materials in mice. Int Wound J. 2015;12:122–131.
- Haraga I, Abe S, Jimi S, et al. Increased biofilm formation ability and accelerated transport of Staphylococcus aureus along a catheter during reciprocal movements. J Microbiol Methods. 2017;132:63–68.
- Periasamy S, Joo HS, Duong AC, et al. How Staphylococcus aureus biofilms develop their characteristic structure. Proc Natl Acad Sci USA. 2012;109:1281–1286.
- Yocum RR, Rasmussen JR, Strominger JL. The mechanism of action of penicillin. Penicillin acylates the active site of Bacillus stearothermophilus D-alanine carboxypeptidase. J Biol Chem. 1980;255:3977–3986.
- Mun SH, Joung DK, Kim YS, et al. Synergistic antibacterial effect of curcumin against methicillin-resistant Staphylococcus aureus. Phytomedicine. 2013;20:714–718.
- Li L-M, Li J, Zhang X-Y. Antimicrobial and molecular interaction studies on derivatives of curcumin against Streptococcus pneumoniae which caused pneumonia. Electron J Biotechnol. 2016;19:8–14.