
Abstract
The biological characteristics and genome-wide sequence analysis of highly efficient endophytic nitrogen-fixing bacteria Klebsiella variicola GN02 isolated from the roots of Pennisetum sinense Roxb. were studied by the combination of second-generation and third-generation sequencing techniques. The cell cultivation characteristics, microscopic morphological observation and infrared spectroscopic analysis of this GN02 strain were performed and its genome assembly, gene prediction and functional annotation, eggNOG/GO clustering analysis and co-linearity analysis were conducted through corresponding software. The results showed that the GN02 strain had the basic morphological characteristics of genus Klebsiella and it had the characteristic absorption peak of marker C-N group and the characteristic absorption peaks of carbonyl and amide groups in nitrogen-fixing bacteria. The GN02 genome size was 5,599,366 bp with GC content of 57.41%, 5,261 ORF, 25 rRNA, 87 tRNA, 125 other ncRNA, 7 CRISPR repeats, and 54 GIs. Gene function annotations indicate that there were a large number of genes closely related to cellular nitrogen metabolism and the genes with high abundance are associated with amino acid metabolism. The genome-wide sequence has been submitted to the GenBank database under the accession number of CP31061. The basic characteristics of GN02 genome are similar to those of other K. variicola. Several strains of K. variicola have good co-linearity. The GN02 strain has a complete nitrogen-fixing gene group and 57 nitrogen-fixing genes have been identified.
Introduction
Endophytic diazotroph is a group of microorganisms that colonize healthy plants and host plants for executing combinatorial nitrogen-fixing function [Citation1]. Currently, all of them are endogenous nitrogen-fixing bacteria, but no endogenous nitrogen-fixing fungi have been reported [Citation2,Citation3]. Since 1980s, endogenous nitrogen-fixing bacteria have been found in Gramineae plants, such as sugarcane [Citation4], rice [Citation5], corn [Citation6], and pasture [Citation7]. Previous studies have shown that these endogenous nitrogen-fixing bacteria not only have stronger nitrogenase activity, but also have the function of promoting plant growth, which has become one of the hot topics in the field of combinatorial nitrogen-fixing research.
Pennisetum sinense Roxb. is a perennial forage grass and belongs to the genus Pennisetum (also known as Giant JUNCAO), with the characteristics of erected and clumped growth, high adaptation, high resistance to multiple castration and high yield [Citation8]. It is introduced to China from South Africa in 2005–2007 by the Institute of Fungi at Fujian Agriculture and Forestry University. Because it is particularly tall during local growth process, it is temporarily named as giant grass, which is identified to be difference from domestic other Pennisetum species so that it is being declared for a new variant. P. sinense has extremely developed root system with the characteristics of obvious main root, thick and deep meat, main root under the ground with the depth of 3–4 m, lateral roots with lateral distribution up to 2.0–2.5 m and very strong drought-resistant capacity. It has strong ecological control function for executing wind-preventing and sand-fixing functions so that it is an energy grass and ecological grass with excellent application potentials [Citation9]. In our laboratory, the high-throughput sequencing analysis of the endophytic nitrogen-fixing bacteria conducted previously has confirmed that the roots of P. sinense at the mature stage are rich in endogenous nitrogen-fixing bacteria, whose species and abundance exceed those in stems and leaves at other growth periods, thereby indicating an important source of endogenous nitrogen-fixing bacteria [Citation10]. A strain, GN02, with strong nitrogen-fixing and growth-promoting properties, has been isolated from P. sinense and identified as Klebsiella variicola.
Klebsiella spp. belongs to the genus Klebsiella in the Enterobacter family. In 2004, Rosenblueth et al. [Citation11] colonized Klebsiella spp. into plants and host plants for providing nitrogen and defined it as K. variicola. After many years’ studies, K. variicola has been confirmed as a common nitrogen-fixing bacterium in plants and is widely distributed in roots and soils of sugarcane, corn, rice and other gramineous plants. It has established a correlation with plants during long-term evolution and development processes and is a microbial resource for the combinatorial nitrogen fixation in the root system [Citation12,Citation13]. Although several strains of Klebsiella are important conditional pathogens and iatrogenic infections, the studies in agricultural microbial and industrial pollution control have shown that Klebsiella itself does not have pathogenicity to plants and it is also a kind of green fungus fertilizer and high-efficiency purifying agent [Citation11,Citation14]. However, it is also believed that the genetic difference between K. variicola strain with combinatorial nitrogen fixation in plants and human Klebsiella spp. strain is small, both have pathogenicity-related factors of K. pneumonia. Therefore, the preparation and application of this kind of microbial fertilizers in the field possess certain risks [Citation15,Citation16].
In our laboratory, K. variicola GN02 isolated from P. sinense has high nitrogenase activity and excellent function of solubilizing phosphorus, secreting phytohormones and antibiotics; therefore, it has potential application as a source of bacterial strains for preparing biological fertilizers [Citation17]. In the present study, GN02 strain was used as the material to analyze the whole genome sequence of K. variicola through combining the second generation sequencing technology based on Illumina MiSeq sequencing platform and the third generation single molecule sequencing technology based on PacBioRS II sequencing platform. Meanwhile, the gene sequence of K. variicola was comparatively analyzed with other plant and animal sources for comprehensive understanding of genetic background of K. variicola, thereby providing a theoretical basis for understanding nitrogen-fixing mechanisms, pathogenic mechanisms and pathogenic risks of K. variicola.
Materials and methods
Bacteria and culture medium
Klebsiella variicola GN02 (CGMCC 1.13619) was isolated from the roots of mature P. sinense and preserved by CGMCC (China General Microbiological Culture Collection Management Center) after identification of colonial morphology, genomic sequencing and the growth promoting property in this study.
Ashby nitrogen-free culture medium: the medium includes: 10 g/L of mannite, 0.2 g/L of KH2PO4, 0.2 g/L of MgSO4·7H2O, 0.2 g/L of NaCl, 0.1 g/L of CaSO4·2H2O, 5.0 g/L of CaCO3, 20 g/L of agar (used with solid medium) and pH 7.0–7.2. Tryptose Soya Agar Media (TSA Media), Maconkey Agar Media (MAC Media), Eosin Methylene Blue Media (EMB Media), Xylose Lysine Deoxycholate Media (XLD Agar) and Blood Agar Base Medium were all bacterial culture medium commonly used in laboratory and were all analytical pure grade, purchased from HOPEBIO Technology (China) Co., Ltd. Luria-Bertani Media: the medium includes 10 g/L of tryptone, 10 g/L of NaCl, 5 g/L of yeast extract and pH 7.0–7.2.
Reagents and instruments
Gram dyeing kit, flagellum dyeing kit and capsule dyeing kit were all purchased from Solarbio Technology (Beijing) Co., Ltd.
Table model high speed centrifuge (Neofuge 13R) was purchased from Shanghai LiShen Scientific Instrument Co., Ltd, thermal cycler (ABI-2720) and sequencer (ABI 3730XL) were purchased from Applied Biosystems Inc. (USA), electrophoresis apparatus (Mini Pro 300 V Power Supply), infrared spectrometer (ThermoFisher Nicolet iS10) and spectrometer (NanoDrop 2000) were purchased from Thermo Electric Company Inc. (USA), scanning electron microscope (Hitachi S-3400N) was purchased from Hitachi (China) Co., Ltd.
Basic biological characteristics of K. variicola GN02
The GN02 strain was inoculated into Ashby, TSA, MAC, EMB, XLD and blood plate medium, and subjected to the inverted cultivation at 30 °C for 48 h. The morphology of the colonies was observed. The basic morphology, capsule and flagella of the strain were observed under a light microscope. The structure of strain GN02 was analyzed by infrared spectrometer through KBr film at the range of 400–4000 cm−1.
Whole genome sequencing of K. variicola GN02
Cell culture and total DNA extraction of the strain: The GN02 strain was inoculated into LB liquid medium and cultivated at 30 °C with a shaking speed of 180 r/min for 24 h. The cells were collected by centrifugation and cell pellets were obtained and washed with PBS buffer (pH 7.2) for three times for future use. The total DNA of the bacteria was extracted by CTAB method to meet the DNA amount of whole genome sequencing. The extraction method was performed according to Sambrook et al. [Citation18]. The primer sequences were designed to be 5′-AGAGTTTGATCCTGGCTCAG-3′ as the forward primer and 5′-CTACGGCTACCTTGTTACGA-3′ as the reversed primer.
Genomic sequencing, assembly, annotation and circle drawing: The WGS (Whole Genome Shotgun) strategy was used for the whole genome sequencing of GN02 strain in this study. First, a library with different inserted fragments was constructed, second generation sequencing technology based on Illumina MiSeq sequencing platform was then combined with the third generation single-molecule sequencing technology. PacBioRS II sequencing platform was used for the whole gene sequencing analysis, which was completed by Shanghai Paisenuo Biotechnology Co., Ltd. The second-generation high-throughput sequencing data was assembled with Kmer-corrected data using A5-miseq V2016825 and SPAdes genome assembler V3.11.1 software to obtain contigs and scaffolds sequences. The three-generation single-molecule sequencing data obtained by Pacbio was used for the assembly through HGAP4 and CANU V1.6 software to obtain the scaffolds sequence. The data from the second and third generation sequencing were spliced to obtain contigs for collinearity analysis through MUMmer V3 software, which was used for the further confirmation of assembly results and positional relationship between contigs, as well as the filling of the gap between contigs. Finally, the results were corrected through pilon V1.22 software, and the final sequence was spliced to obtain the complete sequence [Citation19,Citation20]. The functional sequence analysis, sub-system analysis, subcellular localization analysis and functional annotation of the protein-encoding genes were performed based on the obtained complete sequence according to the previously reported method [Citation21].
Circle diagram drawing: first, the genomic sequence, genome prediction and non-coding RNA prediction information were integrated into a standard GBK (GenBank) format file and cgview software was used to draw the circle diagram of the genome [Citation22]. The image was edited by Photoshop CS. Finally, a complete genome circle map was obtained.
Comparative genomic analysis of K. variicola
The genomic sequence of K. variicola GN02 strain was compared with the genomic sequences of other K. variicola strains with nitrogen-fixing properties, including K. variicola DSM15968, K. variicola DX120E and K. variicola AT22. The genome sequence and annotation information of three strains were downloaded from the GenBank database. The basic characteristics of four strains were counted. Four strains were used to analyze the common genes and specific genes through BLASTp, and the co-linearity analysis of the genome was performed using Mauve software [Citation23]. The program was run with default parameters.
Nitrogen fixation-related gene analysis of K. variicola GN02
According to the genome prediction of K. variicola GN02 whole gene sequence, protein-encoding gene prediction and annotation results and nitrogen fixation-related genes including nitrogenase gene, structural genes and other regulatory genes, ammonium carriers and nitrogen metabolism regulatory genes were analyzed [Citation24] to explore the molecular mechanisms for nitrogen fixation of K. variicola.
Results and analysis
Basic biological characteristics of K. variicola GN02
The GN02 strain could grow on each medium plate with the basically same colony morphology, which exhibited mucus-like round colonies with protrusions. Based on microscopic observation, they were red, thick and short rod-shaped as gram-negative bacteria with a size of 0.5–0.8 × 1–2 μm. Their arrangement was in the alone, double or short chain mode without spores, flagella and movement, but with a capsule or a thick capsule. The GN02 strain had the basic morphological characteristics of the genus Klebsiella.
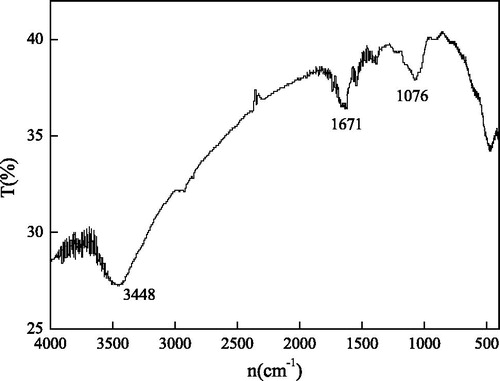
The structural analysis of the strain through infrared spectroscopy was shown in . The characteristic absorption peak of the CN group was at 1076 cm−1, which is the major marker for the identification of nitrogen-fixing bacteria [Citation25]. The absorption peaks at 10,671 and 3448 cm−1 were the symmetric stretching vibration bands of C=O and NH, respectively, indicating the presence of carbonyl and amide groups in nitrogen-fixing bacteria.
Figure 1. IR analysis of GN02 strains. The nitrogen-fixing bacteria have characteristic absorption peaks of CN groups at 1076 cm−1, carbonyl groups at 10671 cm−1 and amide groups at 3448 cm−1.
Whole genome sequence analysis of K. variicola GN02
DNA detection of GN02 strain
The results of agarose gel electrophoresis of DNA samples revealed that high length DNA was successfully isolated, and its electrophoresis bands were single, with no degradation, no protein or RNA contamination. The DNA quality test results showed that the concentration of DNA samples from the strain was 53.20 and 55.10 ng/μL based on fluorescence analysis and ultraviolet analysis, respectively, with a 260/280 ratio of 1.93 and a 260/230 ratio of 1.84. The total volume was 50 μL and total amount was 2.66 μg. The quality evaluation grade was Grade B, and the sample quality was qualified so that it meets the requirements of sequencing library construction.
Genomic assembly and annotation. Sequencing and assembly of genomic sequences
Total reads 2,592,898, total bases 648,487,096 bp, HQ Reads 2,516,158 and HQ Reads 597,933,467 bp were obtained from the second-generation high-quality sequencing, of which HQ Date Reads accounted for 97.04%, HQ Data accounted for 92.20%, and coverage was 72×. Total sequence number of 98, 410, N rate of 0, total sequence length of 1,008,035,098 bp and GC content of 56.75% were obtained from the third-generation sequencing. The sequences from the three-generation sequencing were large in length and had no uncertain bases, which revealed more obvious advantages than the second-generation sequencing. After sequence splicing, the total length of GN02 strain was 5,599,366 bp and the GC Content was 57.41%. The complete sequence is provided as Supplemental Data.
Functional annotation analysis
The protein-encoding genes of GN02 strain were predicted to have a total ORF number of 5,261, ORF total length of 4,867,812 bp and GC content of 58.70%. The ncRNAs were predicted to have 9 sRNAs with total length of 998 bp, 8 sRNAs with total length of 12,288 bp, 23 sRNAs with total length of 23,216 bp, 87 tRNAs with total length of 6,819 bp and 125 other ncRNAs with total length of 16,810 bp. CRISPR analysis predicted a total of seven sequences with confirmed type and total length of 954 bp.
Sub-system analysis
Prophage prediction revealed that GN02 strain had two complete prophages with total length of 100,868 bp, 1 non-complete prophage with length of 17,955 bp and 1 controversial prophage with length of 31,821 bp. GI prediction revealed 54 GIs and the 49th GI was the longest one located at 5,071,887–5,161,760 in the genome, with the size of 89,873 bp (). VFDB analysis revealed 10 virulence genes, such as fepA, fepB, fepC, fepD, fepG, msbA, entA, IlpA, sodB, and iroN, which were the major genes involved in membrane protein transport in combination with iron (). Compared with the K. pneumonia HS11286 strain isolation from clinical, virulence factors related to the pathogenicity of K. pneumonia were not found in GNO2 strain, such as icl, htpB, clpP, ybtA, ybtX, ybtQ, ybtP, and so on [Citation26,Citation27]. The deletion of these virulence factors indicated that GN02 strain was not strong enough to survive in host, resist neutrophil phagocytosis, spread and metastasis of infection. CARD analysis had found 83 antibiotic resistance genes accounting for 1.579% of all protein-encoding genes; 23 antibiotic target genes accounting for 0.437%; and four antibiotic biosynthesis senses accounting for 0.076% related to thiamine synthesis as one of the synthetic genes of acetolactate synthase. CAZy analysis revealed 39 GT, 3 PL, 25 CE, 12 AA, 5 CBM and 78 GH.
Figure 2. GIs forecast in the genome. Red represents the predicted results of the gene island after the integration of the four methods and total 54 GIs were predicted. Blue represents the results predicted by the IslandPath-DIMOB method; yellow, the SIGH-HMM method; green, the IslandPick method and light blue, the Islander method.
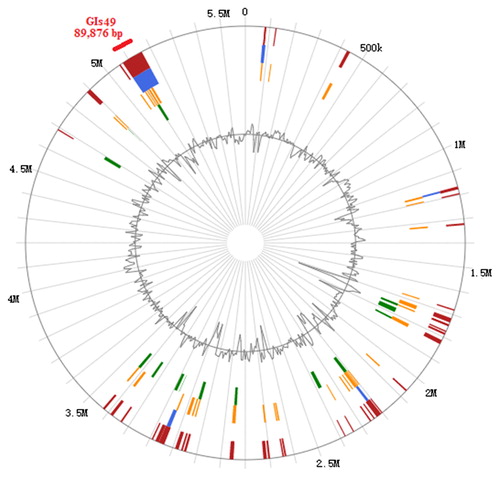
Table 1. Comparison of virulence factors between K. variicola GN02 strain and K. pneumonia HS11286 strain.
Subcellular localization analysis of protein-encoding genes
There were 435 protein-encoding genes in the GN02 genome sequence, which contained 8.27% of total protein-encoding genes. Among them, there were at least one or more trans-membrane helix regions in the 1251 protein-encoding genes, accounting for 23.78% of total protein-enencoding genes. In addition, 346 protein-encoding genes had secretion protein sequences, accounting for 6.58% of total protein-encoding genes.
Function annotation of protein-enencoding genes
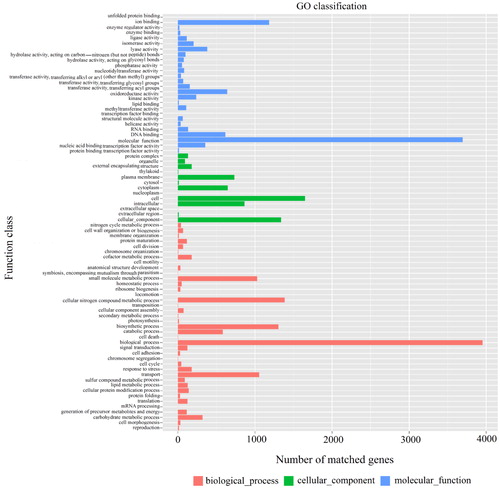
During the comparison from different databases, there were 5,173 protein-encoding genes in the NR database, 4,908 protein-encoding genes in the eggNOG database, 3,226 protein-encoding genes in the KEGG database, 4,491 protein-encoding genes in the Swiss-Prot database, and 4,189 protein-encoding genes in the GO database. Among them, the eggNOG classification annotation indicated that 93.29% protein-encoding genes could be annotated to eggNOG, but the number of unknown function genes was the highest and up to 1,183, accounting for 22.49% of all protein-encoding genes. Among these known functional genes, the genes for regulating carbohydrate transport and metabolism were the most, which were 490 genes accounting for 9.31%, and followed by 460 genes for regulating amino acid transport and metabolism, accounting for 8.74%. The KEGG annotation indicated that the gene abundance associated with membrane transport, carbohydrate metabolism and amino acid metabolism was the highest from the perspective of metabolic pathways. The GOSlim (Gene Ontology Slim) annotation () indicated that the gene functions of the GN02 strain covered all aspects of cellular metabolism, with the most diverse gene types and number associated with biological processes, followed by the genes associated with molecular functions and the genes associated with cellular components. Among these genes related to biological processes, the number of the genes associated with cellular metabolic processes of nitrogen-containing compounds was high and up to 1387, indicating that GN02 strain was closely related to cellular nitrogen metabolism.
Figure 3. Results of GN02 strain genome GOSlim annotation.
Genomic circle drawing
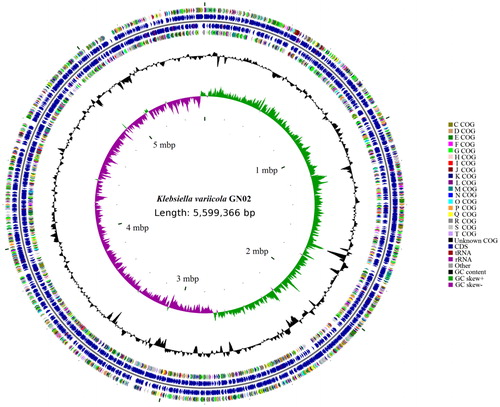
The genomic circle map of GN02 strain was shown in . The whole genome sequence including a circular chromosome with a total length of 5,599,366 bp was finally obtained. After functional annotation of the above gene, it has been registered in NCBI GenBank (CP31061).
Figure 4. GN02 genome circle. From inside to out, the first circle represents the scale, the second circle represents GC Skew, the third circle represents GC content, the fourth and seventh circles represent the COG (Cluster of Orthologous Groups of proteins) to which each CDS (Coding sequence) belongs and the fifth and sixth circles represent the position of CDS, tRAN, and rRAN in the genome.
Comparative genomic analysis of K. variicola
The homology of the four strains of K. variicola was above 99%, and the basic characteristics of four K. variicola genomes were shown in . The GN02 strain had similar genome size, GC content, protein-encoding sequence, average CDS size and the number of RNA genes with other different sources of K. variicola, indicating that GN02 strain had typical characteristics of K. variicola. However, no plasmid was found in GN02 strain and its encoding region percentage was lower than other K. variicola strains, indicating that GN02 strain isolated from P. sinense may have gene loss or acquire some new genes during evolutionary process. Using BLASTp to compare all encoded genes in GN02 strain with other three strains, 4,503 genes were shared by 4 strains, and 427 genes were unique to GN02 strain (), including nitrogen-regulatory protein, exodeoxyribonuclease, sulfatase-modifying factor, hypothetical protein and phage repressor protein, which were transcriptional regulatory proteins, enzymes, unknown proteins, and other relevant proteins. These unique genes provided the unique genetic characteristics of GN02 strain.
Figure 5. Venn diagram of the protein-coding genes of four K. variicola strains. The strains had 4503 common protein-coding genes, while GN02 train had 427 specific protein-coding genes.
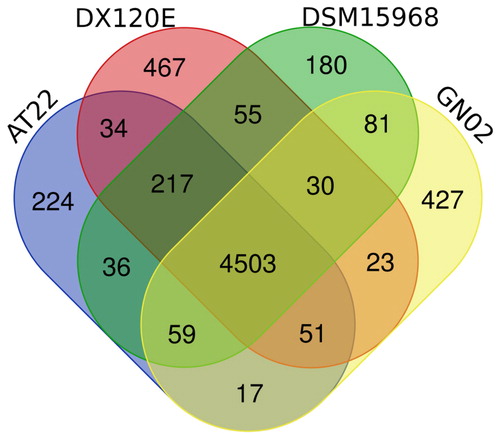
Table 2. General features of the genomes of five K. variicola strains.
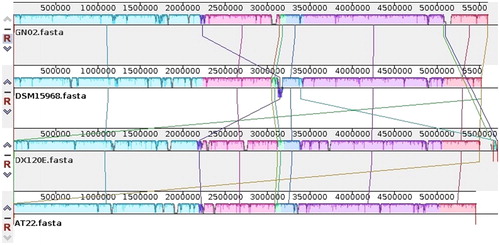
The whole genomes of four K. variicola strains were analyzed by Mauve software (). The results showed that the genomes had good co-linearity and there were five locally co-linear blocks (LCBs). There were some gene insertion and deletion and a small number of inversion, translocation, and genomic rearrangement events in four K. variicola strains.
Figure 6. Synteny block of K. variicola genomes. The same color module represents the collinear region. Good co-linearity of the GN02 strain and other K. variicola strains and five locally co-linear blocks (LCBs) are shown.
Analysis of nitrogen-fixing genes in GN02 strain
Through the whole genome scanning of GN02 strain, the statistical analysis of nitrogen-fixing genes was as follows: 57 nitrogen-fixing genes were identified, including five nif structural genes and 12 cofactors (FeMoco), four nitrogen-fixing enzyme regulatory genes (nifA, nifL), nine ferromoferrin processing, assembly and maturation genes (nifS, nifU, nifM) and 19 other nif genes, as well as seven nitrogen metabolism regulatory genes (ntrB, ntrc, glnD) and one ammonium carrier gene (amtB). The basic information analysis of each nitrogen-fixing gene was shown in . Therefore, GN02 strain has a complete nitrogen-fixing gene group.
Table 3. Analysis of genes related to nitrogen fixation in the GN02 strain.
Final remarks
In recent years, many scholars have reported that K. variicola can be colonized in plants such as sugarcane [Citation28], corn [Citation29], and sarcophagus [Citation30], and has excellent nitrogen-fixing and growth-promoting properties, but the major research focuses on the isolation, identification and growth-promoting of the strains. The molecular mechanisms for nitrogen fixation and growth promotion still need to be further explored.
The activity of nitrogen metabolism such as protein and amino acid synthesis are the most basic metabolic activity of bacterial cells. The GOSlim annotation indicates that the gene abundance related to nitrogen metabolism in the genome of strain GN02 was as high as 1387, ranking fourth among all gene types. This indicated that strain GN02 is closely related to nitrogen metabolism, and there are indeed a large number of genes of this strain involved in nitrogen metabolism, including amino acid synthesis. The eggNOG classification annotation indicates that transport and metabolism of carbohydrates and amino acids account for the highest proportion in the known gene function classification, indicating that GN02 strain is involved in the symbiotic process in host plants, and the relevant metabolic activity of the strain requires the host plant to provide sufficient nutrients [Citation31]. The genes with unknown function have the highest proportion in the gene functional classification, indicating the enormous potentials for discovering new functional genes in GN02 strain. The genome-wide sequencing of GN02 strain has also found a number of genes related to the nitrogen fixation, the secretion of auxin, the dissolution of inorganic phosphorus and iron carriers in plants, such as nif gene cluster (nifHDK, nifLA), nitrogen metabolism-regulatory genes (ntrB, ntrc, and glnD), ammonium carrier gene (amtB), indole-3-pyruvate decarboxylase gene (ipdC), iron cell enteromycin synthesis gene (entABCDEF) and coenzyme PQQ synthesis protein gene (pqqBCDEF). In addition, the prediction of GIs revealed that there are a large number of GIs in the GN02 genome, and the symbiotic characteristics, antagonistic properties and antibiotic secreting genes of the strains are highly associated with GIs [Citation32]. Therefore, an in-depth analysis and exploration of the genomic information of GN02 strain can provide great benefits to understand the colonization model of endophytic nitrogen-fixing bacteria in plants and to explore the genes for regulating the metabolic pathways associated with growth promotion of plants. This useful information can provide a theoretical basis for a better understanding of the interaction between endogenous nitrogen-fixing strains and host plants.
In the present study, a comparative genomic analysis of several K. variicola strains with nitrogen-fixing function was carried out. The genomes of K. variicola from different sources were similar and revealed the excellent co-linearity, indicating that K. variicola has a relatively conservative genomic structure among different hosts. It is speculated that the genetic differences between different strains may be individually different in gene level transfer associated with mobile genetic elements [Citation33]. Some K. variicola strains, such as DX120E derived from sugarcane contain multiple plasmids that are very similar to the plasmids in strains, such as K. pneumoniae and K. oxytoca [Citation30], which has more than 96% homology as the pathogenic plasmids or drug-resistant plasmids. The presence of these plasmids has gained extensive concerns about the potential pathogenicity of K. variicola to animals and plants. Compared with the K. pneumonia strain isolation from clinical, the pathogenicity of GN02 strain is not strong, and these pathogenic or drug-resistant plasmids have not been found in GN02 strain, indicating the reduced possible risks of its application and the great application potential of GN02 strain in agricultural development. It can be used as a strain resource to prepare nitrogen-fixing bacterial fertilizer to promote crop growth and improve crop productivity, which will play an important role in the sustainable development of agriculture in the future.
Conclusion
In the present study, the biological characteristics and genome-wide sequence analysis of a highly efficient endophytic nitrogen-fixing strain K. variicola GN02 isolated from the roots of Phytophthora showed that GN02 strain had basic morphological characteristics of genus Klebsiella and had a nitrogen-fixing bacterial markers, such as characteristic absorption peaks of CN, carbonyl, and amide groups. The complete genome sequence of GN02 strain as a circular chromosome including 5,599,366 nucleotides with 57.41% GC content, 5,261 ORF, 25 rRNA, 87 tRNA, 125 other ncRNA, 7 CRISPR repeats, and 54 GIs was obtained by the combinatorial method of the second and third generation sequencing technology. Gene function annotations revealed a large number of genes in the genome closely related to cellular nitrogen metabolism and high gene abundance associated with amino acid metabolism. The genome-wide sequence had been submitted to the GenBank database under the accession number of CP31061. Comparative genomic analysis showed that the basic characteristics of GN02 genome were similar to those of other K. variicola, and several strains of K. variicola had a good co-linear relationship. The GN02 strain had a complete nitrogen-fixing gene group and 57 identified nitrogen-fixing genes, including nif structure genes and cofactors, nitrogen-fixing enzyme regulatory genes, nitrogen metabolism-regulating genes and ammonium carrier (amtB) genes.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Liaqat F, Eltem R. Identification and characterization of endophytic bacteria isolated from in vitro cultures of peach and pear rootstocks. Biotech. 2016;6:1–8.
- Baldani J, Caruso L, Baldani VLD, et al. Recent advances in BNF with non-legume plants. Soil Biol Biochem. 1997;29:911–922.
- Purchase BS. Nitrogen fixation associated with sugarcane. Congr S Afr Sugar Technol Assn. 1980;137:173–176.
- Martínez-Romero E, Silva-Sanchez J, Barrios H, et al. Draft genome sequences of Klebsiella variicola plant isolates. Genome Announc. 2015;3:2. DOI: 10.1128/genomeA.01015-15
- Pham VT, Rediers H, Ghequire MG, et al. The plant growth-promoting effect of the nitrogen-fixing endophyte Pseudomonas stutzeri A15. Arch Microbiol. 2017;199:513–517.
- Szilagyi-Zecchin VJ, Ikeda AC, Hungria M, et al. Identification and characterization of endophytic bacteria from corn (Zea mays L.) roots with biotechnological potential in agriculture. AMB Exp. 2014;199:1–9.
- Dalton DA, Kramer S, Azios N, et al. Endophytic nitrogen fixation in dune grasses (Ammophila arenaria and Elymus mollis) from Oregon. FEMS Microbiol Ecol. 2004;49:469–479.
- Wang YS, Zhou BF. The sociological value of invention and development of Juncao technology. J Fujian Agric for Univ. 2006;9:1–5. Chinese.
- Lin XS, Lin ZX, Lin DM, et al. Effects of planting Pennisetum sp. (Giant juncao) on soil microbial functional diversity and fertility in the barren hillside. Acta Ecol Sin. 2014;34:304–4312. Chinese.
- Lin BS, Fan JL, Song ZZ, et al. Endophytic diazotrophs composition of Pennisetum sp. at different growth stages. Microbiol China. 2018;45:1479–1490.
- Rosenblueth M, Martínez L, Silva J, et al. Klebsiella variicola, A novel species with clinical and plant-associated isolates. Syst Appl Microbiol. 2004;27:27–35.
- Zhang KK, Xing YX, Shao M. Cloning, prokaryotic expression and proteinpurification of sugarcane endogenous azotobacter klebsiella variicola DX120 Enifk gene. Int Res J Nat Appl Sci. 2015;2:69–85.
- Rodríguez-Blanco A, Sicardi M, Frioni L. Plant genotype and nitrogen fertilization effects on abundance and diversity of diazotrophic bacteria associated with maize (Zea mays L.). Biol Fertil Soils. 2015;51:391–402.
- Li MJ, Peng S, Xu SZ, et al. Application of Klebsiella spp. in agriculture and environmental management. Curr Biotechnol. 2014;4:415–420. Chinese.
- Long SW, Linson SE, Ojeda Saavedra M, et al. Whole-genome sequencing of human clinical Klebsiella pneumoniae isolates reveals misidentification and misunderstandings of Klebsiella pneumoniae, Klebsiella variicola, and Klebsiella quasipneumoniae. Msphere. 2017;2:15. DOI: 10.1128/mSphereDirect.00290-17
- Reyna-Flores F, Barrios-Camacho H, Dantán-González E, et al. Draft genome sequences of endophytic isolates of Klebsiella variicola and Klebsiella pneumoniae obtained from the same sugarcane plant. Genome Announc. 2018;6:2. DOI: 10.1128/genomeA.00147-18
- Lin BS, Wang LF, Zhang KL, et al. Study on effect of interaction between endophytic diazotrophs from Pennisetum sp. and P-releasing bacteria and manufacture of their compound bacterial fertilizer. Jiangsu Agric Sci. 2018;46:218–283. Chinese.
- Sambrook J, Russell D. Molecular cloning: a laboratory manual. 3rd ed. New York: Cold Spring Harbor Laboratory Press; 2016.
- Delcher AL, Kasif S, Fleischmann RD, et al. Alignment of whole genomes. Nucleic Acids Res. 1999;27:2369–2376.
- Walker BJ, Abeel T, Shea T, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:14. DOI: 10.1371/journal.pone.0112963
- Liu J, Zhao Z, Deng Y, et al. Complete genome sequence of Vibrio campbellii LMB 29 isolated from red drum with four native megaplasmids. Front Microbiol. 2017;8:1–13.
- Stothard P, Wishart DS. Circular genome visualization and exploration using CGView. Bioinformatics. 2005;21:537–539.
- Darling AC, Mau B, Blattner FR, et al. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–1403.
- Li QJ, Cheng JJ. Sun SX. Isolation, identification and characterization of associative nitrogen-fixing endophytic bacterium Kosakonia radicincitans GXGL-4A in maize. Microbiol China. 2016;43:2456–2463.
- Dai QW, Dong FQ. The application study of FTIR technology in microbial classification. J Southwest Univ Sci Technol. 2009;24:114–118. Chinese.
- Turton JF, Perry C, Elgohari S, et al. PCR characterization and typing of Klebsiella pneumoniae using capsular type-specific, variable number tandem repeat and virulence gene targets. J Med Microbiol. 2010;59:541–547.
- Chou HC, Lee CZ, Ma LC, et al. Isolation of a chromosomal region of Klebsiella pneumoniae associated with allantoin metabolism and liver infection. Infect Immun. 2004;72:3783–3792.
- Wei CY, Lin L, Luo LJ, et al. Endophytic nitrogen-fixing Klebsiella variicola strain DX120E promotes sugarcane growth. Biol Fertil Soils. 2014;50:657–666.
- Marag PS, Suman A. Growth stage and tissue specific colonization of endophytic bacteria having plant growth promoting traits in hybrid and composite maize (Zea mays L.). Microbiol Res. 2018;214:101–113.
- Zhao MF, Li MJ, Zhang FF, et al. Colonization of Klebsiella variicola in dendrobium officinale and its control effect on dendrobium alternaria. J Southwest For Univ. 2015;35:14–19. Chinese.
- Lin L, Wei C, Chen M, et al. Complete genome sequence of endophytic nitrogen-fixing Klebsiella variicola strain DX120E. Stand Genomic Sci. 2015;10:1–7.
- Shay JA, Dhillon BK, Laird MR, et al. IslandViewer and IslandPlot: flexible identification, visualization, and characterization of microbial genomic regions associated with adaptations of environmental, medical, and industrial importance. Genome. 2014;57:394–394.
- Chen M, Li Y, Li S, et al. Genomic identification of nitrogen‐fixing Klebsiella variicola, K. pneumoniae and K. quasipneumoniae. J Basic Microbiol. 2016;56:78–84.