
Abstract
Drought is a major factor that influences the productivity of crops including wheat. However, drought tolerance is a complex trait involving many genes and cellular pathways. To help understand the complex regulatory mechanisms governing drought tolerance in wheat, we performed small RNA sequencing analysis in a highly drought-tolerant wheat variety (Triticum aestivum L. cv. XF 20). Experimental plants were cultivated in the presence of PEG6000 to induce drought stress, while control plants were grown in the absence of PEG6000. Sequencing results confirmed the expression of 199 previously reported miRNAs. We also identified 32 novel miRNAs in this species. The expression of 37 miRNAs was significantly affected by PEG6000-induced drought stress (27 upregulated, 10 downregulated), including 5 novel miRNAs. Degradome sequencing showed that 222 target gene mRNAs were cleaved by these miRNAs. The target genes identified by psRNATarget and degradome were all enriched in gene ontology categories. There were significant differences in the number of target genes regulated by differentially expressed miRNAs, especially in signal transducer, response to stress and antioxidant pathways. MiRNA targets that are known to be related to drought and/or stress response in plants included 6-phosphogluconate dehydrogenase, epoxide hydrolase, sucrose synthase and phytochrome and flowering time regulatory protein. Our results shed light on the molecular mechanisms of drought tolerance in wheat, and identify drought-responsive miRNAs and their targets in a drought-resistant variety. These results could be exploited to improve wheat and related plants, which is expected to be vital as agriculture addresses the consequences of climate change.
Introduction
Plants have developed remarkable capabilities to adapt to extreme environments, such as high temperature, drought and salt stress [Citation1]. Drought is a common environmental stress, and dry weather is becoming more frequent with intensification of the greenhouse effect and climate change [Citation2, Citation3]. This is particularly true in the agricultural areas of northwestern China, which are frequently exposed to drought conditions and dry, hot winds. Drought limits the growth, development and ultimately, the yields of crop plants worldwide [Citation4]. Plants respond to stresses by regulating the expression of specific genes to avoid or minimize cellular damage, thus adapting to stress conditions [Citation5].
Recently, the discovery of microRNAs (miRNAs) revealed a new and additional layer of post-transcriptional gene regulation. MiRNAs are 21 to 24 nucleotides (nt) long, and are noncoding RNAs that can negatively regulate gene expression [Citation6–8]. Plant miRNAs are highly complementary to target messenger RNAs (mRNAs), and miRNA sequences are generally highly conserved between related organisms. Increasing evidence indicates that miRNAs participate in a diverse set of processes such as plant growth, signal transduction, apoptosis, and biotic and abiotic stress responses [Citation9–13]. Drought-stress-related miRNAs have been identified by small RNA sequencing technology in rice [Citation14], wheat [Citation15], sugarcane [Citation16], Medicago truncatula [Citation17], and potato [Citation18]. Upon drought stress, approximately 14 miRNAs were found to be significantly differentially expressed in bread wheat [Citation2], while expression patterns and response varied between root and leaf [Citation19]. One miRNA may regulate many target genes [Citation20, Citation21], and the biological functions of miRNAs can be deduced by identification and characterization of their target genes. To this end, characterization of the degradome typically includes deep sequencing, bioinformatic analysis, and 5′-rapid amplification of cDNA ends. Degradome analysis has been used for global identification of miRNA–target RNA pairs in numerous plant species, including Arabidopsis [Citation22], rice [Citation23], wheat [Citation24], soybean [Citation25], maize [Citation26] and grapevine [Citation27].
Hexaploid wheat (Triticum aestivum L. AABBDD) is a major food crop, nurturing more than one-third of the world’s population and providing nearly 55% of carbohydrates grown and consumed [Citation28, Citation29]. Environmental constraints, such as high temperature (or heat stress), drought, and the combination of the two, severely and antagonistically limit wheat yield and quality [Citation5, Citation7–9]. It is predicted that a global temperature increase of 2 °C above current averages could lead to wheat yield reductions of up to 50% via perturbations in physiological and biochemical processes [Citation30]. Moreover, drought is reported to adversely affect >50% of current global wheat cultivation land, causing considerable yield loss through the inhibition of photosynthesis [Citation6, Citation31].
In this work, we investigated the differential expression of miRNAs in the wheat cultivar XF 20 in response to drought stress, and identified targets of these miRNAs. We also identified 32 novel miRNAs in wheat. As this variety of wheat is drought-resistant, knowledge of its regulatory and biochemical mechanisms in response to drought may be used in programmes to improve the resistance of other varieties of wheat.
Materials and methods
Plant growth and treatment
The XF 20 wheat cultivar has been used in the identification of factors influencing drought resistance in a national regional trial and was first released by Qingyang Academy of Agricultural Sciences in 1984 [Citation32]. This variety has grade 2 drought resistance, and it is mainly resistant to heat and drought, hot wind (National Infrastructure of Plant Germplasm Resources, Beijing). Supplementary information Table S1 shows the indices of different wheat varieties under drought stress—the drought-resistant phenotype of Xifeng 20 is obvious.
For our experiments, seeds of wheat cultivar XF 20 were surface-sterilized in 1% sodium hypochlorite for 20 min, rinsed in distilled water six times, and soaked in the dark at room temperature overnight. All seedlings were incubated in a growth chamber [24 °C/18 °C (day/night), 14 h/10 h (light/dark), 60% humidity]. The seedlings were irrigated with half-strength Hoagland solution [Citation33] once every other day, and after 21 days they were randomly divided into two groups. For drought stress (treatment group), seedlings were treated with 20% (w/v) PEG-6000 for 6 h (roots were totally covered). The untreated group (control) was incubated as above without exposure to PEG-6000. All experiments were performed in parallel. Roots were collected 6 h after the start of treatment. For the two groups (control and treatment), the roots of six randomly chosen seedlings were pooled to form a biological replicate, snap frozen in liquid nitrogen and stored at −80 °C until further use.
Small RNA library construction and sequencing
Small RNAs were extracted from plant samples from the two groups (control and drought) using TRIzol reagent (Invitrogen, Carlsbad, CA), according to the manufacturer’s instructions. Small RNAs were ligated sequentially to 59- and 39-RNA/DNA chimeric oligonucleotide adaptors. The resulting ligation products were gel-purified by 15% denaturing polyacrylamide gel electrophoresis, and reverse-transcribed to produce cDNAs. cDNAs were sequenced using a Genome Analyzer IIx System (Illumina, USA), according to the manufacturer’s instructions.
Identification of conserved and novel miRNAs
Raw data were first processed by filtering out low-quality reads, trimming the adaptors and removing other noisy reads, to obtain clean reads. Clean reads were aligned to the Rfam database to remove other noncoding RNAs, including rRNAs, tRNAs and snRNAs. The remaining reads were mapped to assembled transcriptome sequences from Brachypodium distachyon [Citation34]. Mapped reads were retrieved and aligned to plant miRNA sequences from miRBase V21 [Citation5] using Bowtie [Citation35], which identified and annotated conserved miRNA genes. To identify novel miRNA genes, first, miRDeep-P retrieved the flanking assembled transcriptome sequences of mapped reads, which were identified as candidate precursors of miRNAs [Citation36]. Second, based on the precursors, novel miRNAs were identified using MIREAP, and their secondary structures were verified by the software RNAfold, as previously reported [Citation37]. In addition, plant miRNA criteria for Arabidopsis [Citation38] and rice [Citation39] were also considered for novel wheat miRNA identification.
All miRNA abundances were evaluated and normalized using the tags per million reads (TPM) method, based on BLAST mapping results. The TPM values were calculated as: TPM = (number of mapped miRNA reads × 106)/number of clean sample reads. The normalised expression was adjusted to 0.01 when miRNA expression (TPM) was zero to avoid negating subsequent calculation of fold-change. The miRNA expression fold-changes between drought-stress and control group plants were computed, and chi-square tests were performed to determine the significance of miRNA expression for each comparison using R software. The miRNAs with fold change (TPM ratios) ≥2 or ≤0.5, and p value ≤0.05, were deemed to be differentially expressed in response to drought stress, as described by Xie et al. [Citation40].
Degradome library construction and target identification
To investigate the potential targets of miRNAs, a degradome library was constructed from drought stress samples, as previously described [Citation41]. In brief, poly(A)-enriched RNAs were isolated, and ligated to an RNA oligonucleotide adaptor containing a 39-MmeI recognition site; the ligated products were used to synthesize first-strand cDNA. A short PCR program (five cycles) was used to amplify the cDNA, and the product was ligated to a double-stranded DNA adaptor before being subjected to gel purification again for PCR amplification. The final cDNA library was purified and sequenced on an Illumina Genome Analyzer IIx by BGI-Shenzhen Co. Ltd.
Adaptor sequences and low-quality sequencing reads were removed from the raw reads, and the clean reads were used to identify potentially cleaved targets based on B. distachyon assembled transcriptome sequences (described below) by the CleaveLand4 pipeline [Citation42]. The psRNATarget tool was also used to predict miRNA targets using a set of default parameters [Citation43]. MiRNA target genes were also predicted from B. distachyon assembled transcriptome sequences. Prediction targets were used to cross-check the degradome sequencing results.
Data availability
The Triticum aestivum (cv. XF 20) small RNA sequences, degradome sequences, and transcriptome sequences have all been deposited in the NCBI SRA database with accession number SRP 166912.
Results and discussion
High-throughput sequencing of small RNA libraries
To identify novel miRNAs expressed in wheat (cv. XF 20), we successfully produced a total of 11,723,790 and 12,034,102 clean reads from control and drought-treated plant cDNA libraries, respectively (summarized in ). The length of the small RNA reads from the two libraries ranged from 18 to 30 nt. The clean reads were mapped to the Triticum aestivum genome assembly; about 79.9% of control and 84.4% of drought-treated plant reads were mapped. To identify conserved miRNAs, the mappable reads were aligned to known plant miRNAs in the miRbase database, using Bowtie with no more than one mismatch. In total, 814,254 and 1,287,494 reads from the control and drought libraries, respectively, were identified as homologous to known miRNAs. The flanking sequences of the remaining mappable reads were retrieved and analyzed by MIREAP using plant default parameters. In total, 32 novel miRNAs were identified in XF 20, most being 21 and 24 nt long (Supplementary information Table S2).
Table 1. Overview of small RNA sequences for two wheat (Triticum aestivum L. cv. XF 20) libraries.
To identify miRNAs involved in the response of XF 20 plants to drought stress, we evaluated and analyzed the miRNA expression in the roots of both the control and drought plant groups. Compared with the control group, miRNAs with a log2 fold-change >1, or < −1, combined with a p value <0.05, were identified as differentially expressed miRNAs. We identified 37 significantly differentially expressed miRNAs in response to drought stress (). Most (27/37) were downregulated by drought stress; ten ones (10/37) were upregulated. Five of the 37 differentially expressed miRNAs were novel miRNAs, two (upregulated) and three (down-regulated) predicted novel miRNAs were possibly related to drought resistance in wheat.
Table 2. Differentially expressed miRNA genes in wheat (Triticum aestivum L. cv. XF 20) in response to drought stress.
Identification of drought-responsive miRNA targets
To determine the function of drought-responsive miRNAs in XF 20 wheat, degradome sequencing was used to identify the miRNA targets. A total of 20,665,443 clean reads were obtained from the degradome cDNA libraries; most were 20- or 21-nt long. The reads were aligned to wheat transcriptome sequences: 17,591,286 reads mapped to the assembled transcriptome sequences. The CleaveLand pipeline was used for identification of target mRNAs; 222 potential target mRNAs that are predicted to be cleaved by known and novel miRNAs were identified from the CleaveLand degradome library analysis (Supplementary information Figure S1). To validate target genes identified through degradome sequencing, miRNA and transcript sequences were submitted to psRNA. There were 90 (90/222, 44.6%) targets from the degradome that were present in the psRNA target results list. These results indicated that degradome sequencing is an effective method to identify cleaved targets of miRNAs.
Analysis of target genes
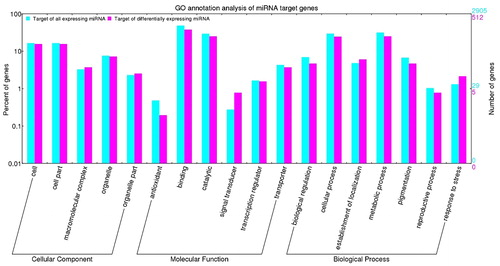
To assess the functions of differentially expressed miRNAs, the target gene list was BLAST searched against a combined database of Arabidopsis, rice and B. distachyon. Considering all expressed miRNAs, a total of 2,905 target mRNAs were identified; 512 of these target mRNAs were targets of differentially expressed miRNAs. These target mRNAs were analyzed by gene ontology (GO) (). Relative to the targets of all expressed miRNAs, GO categories ‘signal transducer’ and ‘response to stress’ showed a higher number of differentially expressed miRNAs than would be expected; the category ‘antioxidant’ contained fewer differentially expressed miRNAs than would be expected. Thus, in drought conditions, differentially expressed miRNAs reduced the inhibition of antioxidant genes, increasing the expression of antioxidant genes to resist drought, while the opposite occurred for signal transduction genes.
Figure 1. Gene ontology analysis of miRNA target genes identified in drought stress of wheat (Triticum aestivum L. cv. XF 20). Note: The digits on the left y-axis and right y-axis show the percentage and number of miRNA targets, respectively. Targets of all expressed miRNAs are shown in cyan; targets of all differentially expressed miRNAs are shown in pink.
Among the identified target genes, many have reported roles in plant responses to drought and other biotic and abiotic stresses. To give examples, we considered genes whose mRNAs were found to be targets of the differentially expressed miRNAs in our CleaveLand degradome library analysis. Novel miRNAs 08 and 15 were identified in this study (Supplementary information Table S2) and were downregulated in wheat cv. XF 20 in response to induced drought stress (). These miRNAs were predicted to target mRNA of genes for disease resistance proteins PRM1-like and RPP13-like. In Arabidopsis, these proteins provide resistance to the bacterial and pathogens Pseudomonas syringae and Hyaloperonospora parasitica (formerly Peronospora parasitica), respectively [Citation44, Citation45]. Notably, NmiRNA15 also targeted mRNA encoding sucrose synthase. Sucrose is the main carbohydrate transported to developing plant organs. In soybean nodules, sucrose synthase was found to decline dramatically in activity and content in response to drought, but the nodule content of sucrose increased [Citation46]. In tall fescue, the sucrose content of leaves increased in response to drought stress [Citation47]. In wheat, it was recently shown that sucrose synthase gene expression was significantly higher in a drought-tolerant cultivar than in a drought-sensitive one. More sucrose accumulated in the tolerant cultivar under drought stress [Citation48]. Similarly, the drought-tolerant cultivar JD17 accumulated sucrose on prolonged drought-stress [Citation49]. Thus, sucrose levels appear to be crucial to the response of wheat to drought, and our results indicate that one response mechanism may involve regulation of sucrose synthase via NmiRNA15.
NmiRNA15 also targeted phytochrome and flowering time regulatory protein (PFT1, also called Mediator 25), a multifunctional nuclear protein involved in response to light and hormone signalling, defence against fungus, and abiotic stress response (including to drought) [Citation50, Citation51]. This protein positively and negatively regulates drought response, for example, through interactions with the transcription factors dehydration responsive element binding protein 2 and ethylene response factor 1 [Citation51].
Among the previously reported miRNAs that were found in this study to be differentially regulated in response to drought-stress (), miR482a-5p has been implicated in the response to pathogens in mulberry trees [Citation52]. MiR482 was downregulated in response to drought in T. aestivum cv. Sivas 111/33 [Citation2]. In our analysis, miR482a-5p, as well as miR9674a-3p, was predicted to target mRNA encoding a protein annotated as a stripe rust resistance protein. Stripe rust is a serious disease of wheat caused by the fungus Puccinia striiformis var. tritici. MiR2111 has been reported to play a role in phosphate homeostasis and plant survival [Citation53]. Here, notably, miR2111c targeted 6-phosphogluconate dehydrogenase (6-PGDH), one of the key enzymes of the oxidative pentose phosphate pathway. In rice, transcript levels and enzyme activity of 6-PGDH were upregulated in response to drought, cold and high salinity [Citation54]. Enhanced 6-GDPH activity has been shown to be related to aluminium tolerance in wheat [Citation55]. MiR5054, which was also downregulated in response to drought stress in our study (), was predicted to target mRNA encoding epoxide hydrolase (EH). EH, which is involved in lipid metabolism, was previously found to be upregulated in a drought-resistant variety of wild emmer wheat (Triticum turgidum ssp. dicoccoides), the progenitor of cultivated wheat [Citation56]. In Arabidopsis, an EH is induced in stems and leaves in response to drought [Citation57].
Conclusions
We report the identification of differentially expressed miRNAs in drought-stressed wheat plants (Triticum aestivum L. cv. XF 20). This variety of wheat is particularly resistant to drought conditions, especially heat and dry wind. Our results shed light on the molecular mechanisms that influence drought tolerance in wheat. Targets of the drought-responsive miRNAs include many factors with well-characterized roles in stress responses, including drought. These miRNAs and their target genes may be exploited via breeding programmes or genetic engineering to improve drought tolerance in other varieties of wheat and related plants.
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
References
- Jenks MA, Hasegawa PM. Plant abiotic stress. Oxford, UK: Blackwell; 2005.
- Akdogan G, Tufekci ED, Uranbey S, et al. miRNA-based drought regulation in wheat. Funct Integr Genomics. 2016;16:221–233.
- Ding Y, Tao Y, Zhu C. Emerging roles of microRNAs in the mediation of drought stress response in plants. J Exp Bot. 2013;64:3077–3086.
- Zhu JK. Salt and drought stress signal transduction in plants. Annu Rev Plant Biol. 2002;53:247–273.
- Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucl Acids Res. 2014;42:D68–D73.
- Thomson DW, Dinger ME. Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet. 2016;17:272–283. 2016.20
- Mittler R. Abiotic stress, the field environment and stress combination. Trends Plant Sci. 2006;11:15–19.
- Zhang B. MicroRNA: a new target for improving plant tolerance to abiotic stress. J Exp Bot. 2015;66:1749–1761.
- Chen X. Small RNAs and their roles in plant development. Annu Rev Cell Dev Biol. 2009;25:21–44.
- Puzey JR, Karger A, Axtell M, et al. Deep annotation of Populus trichocarpa microRNAs from diverse tissue sets. PLoS One. 2012;7:e33034.
- Chen L, Ren YY, Zhang YY, et al. Genome-wide profiling of novel and conserved populus microRNAs involved in pathogen stress response by deep sequencing. Planta. 2012; 235:873–883.
- Qin Y, Duan Z, Xia X, et al. Expression profiles of precursor and mature microRNAs under dehydration and high salinity shock in Populus euphratica. Plant Cell Rep. 2011; 30:1893–1907.
- Sunkar R, Li YF, Jagadeeswaran G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012;17:32–41.
- Barrera-Figueroa BE, Gao L, Wu Z, et al. High throughput sequencing reveals novel and abiotic stress-regulated microRNAs in the inflorescences of rice. BMC Plant Biol. 2012;12:132.
- Ma X, Xin Z, Wang Z, et al. Identification and comparative analysis of differentially expressed miRNAs in leaves of two wheat (Triticum aestivum L.) genotypes during dehydration stress. BMC Plant Biol. 2015;15:21.
- Thiebaut F, Grativol C, Tanurdzic M, et al. Differential sRNA regulation in leaves and roots of sugarcane under water depletion. PLoS One. 2014;9:e93822.
- Wang T, Chen L, Zhao M, et al. Identification of drought-responsive microRNAs in Medicago truncatula by genome-wide high-throughput sequencing. BMC Genomics. 2011;12:367.
- Zhang N, Yang J, Wang Z, et al. Identification of novel and conserved MicroRNAs related to drought stress in potato by deep sequencing. PLoS One. 2014;9:e95489. 10.1371/journal.pone.009548919
- Eldem V, Çelikkol Akçay U, Ozhuner E, et al. Genome-wide identification of miRNAs responsive to drought in peach (Prunus persica) by high-throughput deep sequencing. PLoS One. 2012;7:e50298.
- Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610.
- Nazarov PV, Reinsbach SE, Muller A, et al. Interplay of microRNAs, transcription factors and target genes: linking dynamic expression changes to function. Nucleic Acids Res. 2013; 41:2817–2831.
- Addo-Quaye C, Eshoo TW, Bartel DP, et al. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 2008;18:758–762.
- Sun Z, He Y, Li J, et al. Genome-wide characterization of rice black streaked dwarf virus-responsive microRNAs in rice leaves and roots by small RNA and degradome sequencing. Plant Cell Physiol. 2015;56:688–699.
- Chen F, Zhang X, Zhang N, et al. Combined small RNA and degradome sequencing reveals novel miRNAs and their targets in the high-yield mutant wheat strain Yunong 3114. PLoS One. 2015;10:e0137773.
- Shamimuzzaman M, Vodkin L. Identification of soybean seed developmental stage-specific and tissue-specific miRNA targets by degradome sequencing. BMC Genomics. 2012;13:310.
- Zhao Y, Xu Z, Mo Q, et al. Combined small RNA and degradome sequencing reveals novel miRNAs and their targets in response to low nitrate availability in maize. Ann Bot. 2013;112:633–642.
- Pantaleo V, Szittya G, Moxon S, et al. Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J. 2010;62:960–976.
- Samad AFA, Sajad M, Nazaruddin N, et al. MicroRNA and transcription factor: key players in plant regulatory network. Front Plant Sci. 2017;8:565.
- Boucas J, Höpker K, Chen S, et al. Posttranscriptional regulation of gene expression-adding another layer of complexity to the DNA damage response. Front Genet. 2012;3:159.
- Holoch D, Moazed D. RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet. 2015;16:71–84.
- Bartel D. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297.
- Song TX, Li GD. [Breeding report of a new winter wheat variety XiFeng 20]. Gansu Agr Sci Technol. 1995;7:2–3. Chinese.
- Arnon DI, Hoagland DR. Crop production in artificial culture solutions and in soils with special reference to factors influencing yields and absorption of inorganic nutrients. Soil Science. 1940;50:463–485.
- Vogel JP, Garvin DF, Mockler TC, et al. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature. 2010;463:763–768.
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359.
- Yang X, Li L. miRDeep-P: a computational tool for analyzing the microRNA transcriptome in plants. Bioinformatics. 2011;27:2614–2615.
- Denman RB. Using RNAFOLD to predict the activity of small catalytic RNAs. Biotechniques. 1993;15:1090–1095.
- Meyers BC, Axtell MJ, Bartel B, et al. Criteria for annotation of plant MicroRNAs. Plant Cell. 2008;20:3186–3190.
- Jeong DH, Park S, Zhai J, et al. Massive analysis of rice small RNAs: mechanistic implications of regulated microRNAs and variants for differential target RNA cleavage. Plant Cell. 2011;23:4185–4207.
- Xie F, Wang Q, Sun R, et al. Deep sequencing reveals important roles of microRNAs in response to drought and salinity stress in cotton. J Exp Bot. 2015;66:789–804.
- Folkes L, Moxon S, Woolfenden HC, et al. PAREsnip: a tool for rapid genome-wide discovery of small RNA/target interactions evidenced through degradome sequencing. Nucleic Acids Res. 2012;40:e103.
- Addo-Quaye C, Miller W, Axtell MJ. CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics. 2009;25:130–131.
- Dai X, Zhao PX. psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 2011;39:W155–W159.
- Bittner-Eddy PD, Crute IR, Holub EB, et al. RPP13 is a simple locus in Arabidopsis thaliana for alleles that specify downy mildew resistance to different avirulence determinants in Peronospora Parasitica (At). Plant J. 2000;21:177–188.
- Boyes DC, Nam J, Dangl JL. The Arabidopsis thaliana RPM1 disease resistance gene product is a peripheral plasma membrane protein that is degraded coincident with the hypersensitive response. Proc Natl Acad Sci USA. 1998;95:15849–15854.
- Gonzalez EM, Gordon AJ, James C, et al. The role of sucrose synthase in the response of soybean nodules to drought. J Exp Bot. 1995;46:1515–1523.
- Fu J, Huang B, Fry J. Osmotic potential, sucrose level, and activity of sucrose metabolic enzymes in tall fescue in response to deficit irrigation. J Am Soc Hort Sci. 2010;135:506–510.
- Nemati F, Ghanati F, Ahmadi Gavlighi H, et al. Comparison of sucrose metabolism in wheat seedlings during drought stress and subsequent recovery. Biol Plant. 2018;62:595–599.
- Guo R, Shi LX, Jiao Y, et al. Metabolic responses to drought stress in the tissues of drought-tolerant and drought-sensitive wheat genotype seedlings. AoB Plants. 2018;10:ply016.
- Nils E, Davoine C, Benlloch R, et al. The Arabidopsis thaliana Med25 mediator subunit integrates environmental cues to control plant development. Proc Natl Acad Sci USA. 2011;108:8245–8250. https://doi.org/10.1073/pnas.1002981108
- Kazan K. The multitalented mediator25. Front Plant Sci. 2017;8:999.
- Gai YP, Zhao HN, Zhao YN, et al. MiRNA-seq-based profiles of miRNAs in mulberry phloem sap provide insight into the pathogenic mechanisms of mulberry yellow dwarf disease. Sci Rep. 2018;8:812.
- Xu F, Liu Q, Chen L, et al. Genome-wide identification of soybean microRNAs and their targets reveals their organ-specificity and responses to phosphate starvation. BMC Genom. 2013;14:66.
- Hou FY, Huang J, Yu SL, et al. The 6-phosphogluconate dehydrogenase genes are responsive to abiotic stresses in rice. J Integr Plant Biol. 2007;49:655–663.
- Ślaski JJ, Zhang G, Basu U, et al. Aluminum resistance in wheat (Triticum aestivum) is associated with rapid, Al-induced changes in activities of glucose-6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase in root apices. Physiol. Plant. 1996;98:477–484.
- Krugman T, Chagué V, Peleg Z, et al. Differential gene expression in wild emmer wheat genotypes contrasting in drought resistance. In: Appels R, Eastwood R, Lagudah E, et al. editors. Proceedings of the 11th International Wheat Genetics Symposium, Vol. 3; 2008 Aug 24–29; Brisbane, QLD, Australia. Sydney (Australia): Sydney University Press; 2008. p. 910–912.
- Kiyosue T, Beetham JK, Pinot F, et al. Characterization of an Arabidopsis cDNA for a soluble epoxide hydrolase gene that is inducible by auxin and water stress. Plant J. 1994;6:259–269.