
Abstract
Recent studies have debunked the myth about bacterial immortality. Two independent publications, by Stewart et al. (PLoS Biol. 2005;3(2):e45) and Wang et al. (Curr Biol. 2010;20(12):p.1099–1103), provided strong evidence that the two fission products of Escherichia coli K-12, albeit morphologically equivalent, are functionally asymmetric. The growth and the survival rate of one of the cells, the mother cell, are lower than those of the other, the daughter cell. In view of this intriguing finding, bacteria, and in particular E. coli, emerged as a promising model for exploration of the basic principles of aging. It is not yet clear to what extent human and bacterial cells age similarly but at least some signatures of aging, especially at the molecular level, appear to be common. Spontaneous chemical reactions including hydrolysis, oxidation, and glycation (the Maillard reaction) are well known to cause structural and functional deterioration of biomacromolecules. The Maillard reaction, yielding early (Schiff’s bases and Amadori products) and Advanced Glycation End Products (AGEs) on proteins, DNA and amino-lipids, has been long associated with diseases (diabetes, Alzheimer’s, and Parkinson’s diseases) and aging in humans. In previous studies, we have demonstrated that despite the short life span of E. coli, its proteins and chromosomal DNA accumulate both Amadori products and AGEs under normal physiological conditions (Mironova et al. Mol Microbiol. 2005;55(6):p.1801–1811). This article reviews data on the Maillard reaction as a cause of stochastic damage to proteins and DNA implicated in E. coli and human aging.
Introduction
All human beings share an intuitive idea about the meaning of the words ‘aging’ and ‘death’ and scientific definitions can add little to that knowledge. What science is trying to do is to answer the questions ‘why and how do we age and die’ and ‘is it possible to counteract these processes’. The rather philosophic question ‘why do we age’ is best answered by the ‘mutation accumulation’ [Citation1], the ‘antagonistic pleiotropy’ [Citation2] and the ‘disposable soma’ [Citation3] hypotheses of aging. According to these hypotheses and in compliance with Charles Darwin’s theory of evolution [Citation4], mutations, reproduction and natural selection are evolutionary tools elaborated to best adapt the life forms on our planet to its dynamic changes. There are two main types of cells in multicellular organisms, somatic and germ line cells. The somatic cells build up most of the multicellular body, thereby guarding the remaining small portion represented by the germ cells. Only mutations in the germ cells are passed on to the next generations and fixed. Mutations in the somatic cells are not under selection pressure and can accumulate in cells and tissues of adults thus contributing to aging and age-related diseases.
It was long believed that bacteria such as Escherichia coli are immortal, that is, that they are devoid of a natural aging mechanism and that only stochastic events like spontaneous mutations can kill them. A careful look at the old literature, however, shows that deaths among actively proliferating bacteria occur more often than spontaneous mutations. In studies addressing the relevance of the ‘catastrophic error theory’ of senescence, Gallant and Palmer [Citation5] showed that mortality among exponentially growing E. coli varies between one dead cell per 102 to 103 cells, which is quite higher than the average spontaneous mutation rate of one mutant per 108 to 106 cells. Given that not every mutation is deadly, the likelihood that only mutations can kill bacteria equals to zero. It is worth noting that not all unicellular organisms but only those dividing symmetrically were included in the cohort of the immortal beings. Signs of aging were recognized among asymmetrically dividing single-celled organisms. The yeasts Saccharomyces cerevisiae, for instance, reproduce by budding producing a smaller (daughter) cell and a bigger (mother) cell [Citation6,Citation7]. What is the difference between the two cells? The yeast mother cells take on themselves most of the damaged cellular components [Citation8–10] thus relieving the growth of the juvenile daughter cells. Thus, the asymmetric division appears an evolutionary solution to avoid toxic chemical build-up.
Regarding the question ‘how do we age’ most of the aging hypotheses stress on the fact that deterioration of the structure and function of proteins and DNA is crucial to aging. According to the ‘somatic mutation’ [Citation11] and the ‘genome maintenance’ [Citation12] hypotheses, deterioration of DNA repair and accumulation of DNA damage in somatic cells of multicellular organisms significantly contribute to aging. On the other hand, the ‘oxidative damage/free radical’ [Citation13] and the ‘Maillard reaction’ [Citation14,Citation15] hypotheses of aging point to some causes of DNA damage such as oxidation and glycation, respectively. In 2001, we provided evidence that the Maillard reaction, thought to contribute to human aging, also affects the symmetrically diving E. coli [Citation16]. Two later studies, of Stewart et al. [Citation17] and Wang et al. [Citation18], demonstrated that during division E. coli exhibits functional asymmetry in the distribution of some damaged cellular components as a hallmark of aging. Is the Maillard reaction implicated in the asymmetric accumulation of cellular damage in E. coli and aging? The answer to this question and the search for common molecular mechanisms underlying bacterial and human aging are the leitmotifs of this review.
The Maillard reaction
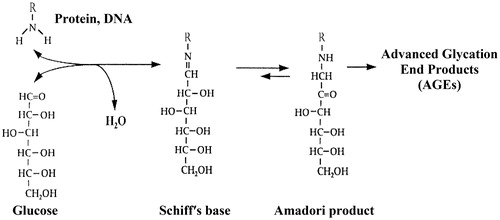
The Maillard reaction is named after the French chemist Louis Camille Maillard, who was first to show that glucose reacts with amino acids in vitro [Citation19]. In the early stage of this reaction, the amino groups of primary amines including proteins and DNA react spontaneously (no catalysis) and reversibly with the carbonyl groups of glucose and other carbonyl compounds to form a Schiff's base (aldimine). Then, at neutral pH, the Schiff's base undergoes an intramolecular rearrangement to a more stable Amadori product (ketoamine) [Citation20]. In the late stage of the Maillard reaction, following additional chemical transformations, the Amadori products give rise to the so-called Advanced Glycation End Products (AGEs) ().
Figure 1. The Maillard reaction.
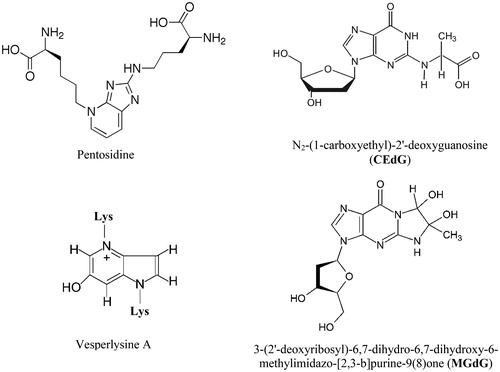
AGEs are an extremely heterogeneous group of covalent adducts, and the chemical structure of a number of AGEs formed in vitro and in vivo has been identified so far [Citation21–28]. Some AGEs form intra- and intermolecular crosslinks and may cause covalent aggregation of affected macromolecules. shows the chemical structure of two AGEs with crosslinking properties promoting protein–protein interactions and aggregation, as well as the structure of two AGEs detected in DNA and nucleotides of human subjects [Citation29,Citation30].
Figure 2. Protein and DNA bound AGEs.
Studies in the late 1960s showed that the Maillard reaction takes place in the human body as well, where it accounts for the formation of a glucose-derived Amadori product at the N-terminus of the haemoglobin β-chain [Citation31]. Glycated haemoglobin (Hbа1c) increases with aging of the red blood cells of healthy subjects and to a higher degree in the erythrocytes of diabetic patients. Because of that Hbа1c is today a routine hyperglycaemic marker. In the mid-1980s, it was hypothesized that glycation plays an important role in the pathogenesis of diabetic complications and aging [Citation14,Citation15]. Subsequently, this hypothesis has found a lot of experimental support and the link between glycation, diabetes and aging has been extensively debated in many reviews [Citation32–37]. The term ‘glycation’ is synonymous to ‘Mailard reaction’ and was introduced in the scientific literature in order to make a clear difference between the enzymatic and the non-enzymatic glycosylation.
Protein glycation in aging and disease
It is proposed that the accumulation of long-lived and stable AGEs in human tissues accelerates the multisystem functional decline that occurs during aging [Citation38]. Although AGEs with both crosslinking and non-crosslinking properties contribute to this process, the current review will focus mainly on the AGEs-promoted protein crosslinking and aggregation as a hallmark of aging. In fact, Johan Bjorksten postulated the role of protein crosslinking in aging [Citation39,Citation40] 10 years before the realization that the Maillard reaction occurs in the human body. Not only healthy aging but also many age-related diseases and diabetic complications are accompanied by protein crosslinking [Citation41,Citation42]. In fact, uncontrolled hyperglycaemia provides a model of accelerated aging helping us to elucidate some aspects of the implication of the Maillard reaction in aging.
Collagen is the main structural protein in the extracellular matrix and the main component of connective tissue, making up roughly 30% of the human whole-body protein. Over 20 collagen types are spread throughout the human body to take part in the architecture of different tissues. Collagen molecules represent triple helices that form higher-order structures (collagen fibrils) through intermolecular crosslinking of lysine residues catalyzed by lysyl-oxidases in the extracellular space. Due to the low turnover of collagen it progressively accumulates AGEs with time including crosslinking AGEs such as pentosidine [Citation43–45] ().
Figure 3. Crosslinking of collagen fibres (cyan) by pentosidine (magenta).
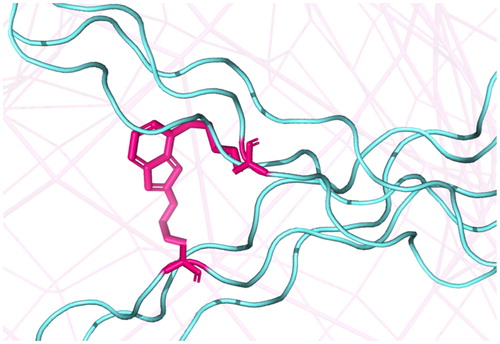
The half-life of cartilage collagen, for example, was calculated to be 117 years and that of skin collagen 15 years, and this half-life was found to correlate positively with the accumulation of pentosidine in the cartilage versus skin collagen [Citation46,Citation47]. While enzymatic intermolecular crosslinking of collagen is essential for its proper functioning, AGEs-crosslinks usually compromise the mechanical properties of collagenous tissues. An increase in collagen crosslinking via pentosidine was shown to correlate with age [Citation48] and to result in bone brittleness [Citation49,Citation50]. Studies with animal models of Type 2 diabetes have also shown that excessive accumulation of pentosidine decreases femoral bone strength [Citation51]. Corneal thickening and stiffness in older people and diabetic patients has been attributed to the accumulation of AGEs-intrinsic fluorescence and pentosidine in corneal collagen [Citation52]. The formation of AGEs-crosslinks in collagen of the arterial extracellular matrix causes arterial stiffness associated with enhanced risk of heart attack [Citation53]. AGEs levels in aorta are significantly elevated by age and diabetes including the AGEs-crosslinks pentosidine and NFC-1. The increase of these two AGEs was shown to correlate positively with aorta stiffness in both control and diabetic subjects [Citation54].
The Maillard reaction contributes also to the pathological transformations of amyloidogenic proteins and peptides including β-amyloid peptide, α-synuclein, and prions associated with neurodegenerative diseases such as Alzheimer’s, Parkinson’s and prion disorders [Citation55,Citation56]. Parkinson’s disease (PD) affects 1–2% of the population aged over 65 years and nearly 4% in those aged above 85 years [Citation57]. The PD phenotype is caused by the loss of dopaminergic mediation in the Substantia nigra pars compacta of the brain's basal ganglia. The most obvious symptoms early in the disease are shaking, rigidity and bradykinesia. In the advanced stages of PD dementia becomes common and is characterized by the formation of Lewy bodies (LBs) in the Substantia nigra pars compacta [Citation58] (). The LBs are composed of cytoplasmic α-synuclein (α-syn) aggregates that displace other cellular components. The aggregation of α-syn is promoted by oxidative and carbonyl stress [Citation56,Citation60] and results in the formation of toxic LBs inclusions and insoluble fibrils as well that disrupt the organization of brain areas responsible for motor control. The interaction between AGEs and their cognate receptors contributes to the generation of a prooxidant tissue environment that appears a significant driver of PD [Citation61]. Dalfó et al. [Citation62] showed that the receptor for AGEs (RAGEs) is expressed in the Substantia nigra, which indicates that AGEs are formed in PD and may have pathological implications. Experiments with α-syn knock-out mice imply that this protein modulates glucose metabolism, as such mice suffer from glucose toxicity and consistent enhancement of certain AGEs. The concomitant up-regulation of glyoxalase I as a protective response was insufficient to prevent AGEs formation [Citation63]. AGEs-crosslinks contribute to α-syn aggregation [Citation64] and in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model α-syn was found to co-localize with well-known AGEs such as Nε-(carboxymethyl)lysine (CML) and Nε-(carboxyethyl)lysine [Citation65,Citation66]. Glycation as a common cause of diabetic complications and α-syn aggregation led some authors to propose that anti-diabetic drugs targeting the levels of glycation may also prove beneficial for treatment of PD [Citation67].
Figure 4. (A) Immunohistochemistry of α-syn showing positive staining (brown) of an intraneural Lewy body in the Substantia nigra in Parkinson’s disease [Citation58]; (B) High magnification micrograph of cerebral amyloid angiopathy with senile plaques in the cerebral cortex consistent of β-amyloid, as may be seen in Alzheimer’s disease [Citation59].
![Figure 4. (A) Immunohistochemistry of α-syn showing positive staining (brown) of an intraneural Lewy body in the Substantia nigra in Parkinson’s disease [Citation58]; (B) High magnification micrograph of cerebral amyloid angiopathy with senile plaques in the cerebral cortex consistent of β-amyloid, as may be seen in Alzheimer’s disease [Citation59].](/cms/asset/153c613c-5f82-4ef2-80f1-41b5097f78f0/tbeq_a_1590160_f0004_c.jpg)
Glycation-promoted crosslinking plays an important role also in Alzheimer’s disease (AD). This rather sporadic neurodegenerative disorder is manifested by progressive loss of cognitive function and affects nearly 5% of people aged 65–75 and almost 50% over 85 [Citation55]. Currently, it was recognized that AD occurs mainly because of aggregation of the extracellular β-amyloid peptide (Aβ) and the intracellular microtubule-associated tau protein. The aggregation of Aβ and tau is contributed mostly by glycation as far as AGEs have been detected in both the Aβ-plaques and neurofibrillary tangles [Citation68,Citation69]. Methylglyoxal (MG) was shown to glycate Aβ and to promote the formation of β-sheets, oligomers and protofibrils and to increase the size of the aggregates [Citation70]. In another study, it was found that the formation of AGEs accelerates the transition from monomeric Aβ to its oligomeric form and then to larger aggregates [Citation71]. Aβ aggregation proceeds through a nucleation-dependent polymerization, which is promoted by crosslinking AGEs. The Aβ aggregation consists of two steps, a first, slow nucleus formation step and a second, rapid elongation step [Citation72]. The first step is reversible and results in oligomer formation. After the oligomers reach a critical size, fast elongation of the aggregates occurs via the addition of Aβ peptides to the ends. The fibril formation is irreversible and the fibrils grow further to form amyloid plaques [Citation59] ().
Glycation damage to DNA and aging
According to the ‘genome maintenance’ hypothesis of aging deterioration of DNA repair and accumulating DNA damage in somatic cells of multicellular organisms are crucial to aging [Citation11,Citation12,Citation73]. Glycation of DNA is expected to impact even more the viability of the organism than protein glycation. As many other lesions, glycation adducts in DNA are rapidly mended by repair, which does not always perfectly restore the original genetic information. Hence, while protein glycation affects the higher order protein structures (secondary, tertiary, and quaternary), DNA glycation may potentially change the amino acid sequence of proteins thus causing gross functional perturbations. Furthermore, because of the complex regulatory networks, mutations in regulatory DNA sequences may be quite more harmful than mutations in a single protein-encoding gene while mutations affecting DNA repair mechanisms themselves are further contributing to excessive DNA damage. Experiments indeed have shown that defects in DNA repair cause premature aging [Citation74–78] and vice versa, enhanced activity of DNA repair enzymes leads to longer life span [Citation79–82]. Many studies reveal a link between the accumulation of mutations in DNA with age and cancer predisposition of elderly people [Citation83–85]. It is unknown yet whether the DNA repair machinery can cope with any type of DNA damage, some evidence suggesting that ‘irreparable DNA lesions’ might exist [Citation86]. Unrepaired DNA lesions lead to replication fork stalling, thereby inflicting cell dysfunction, senescence and apoptosis [Citation87,Citation88].
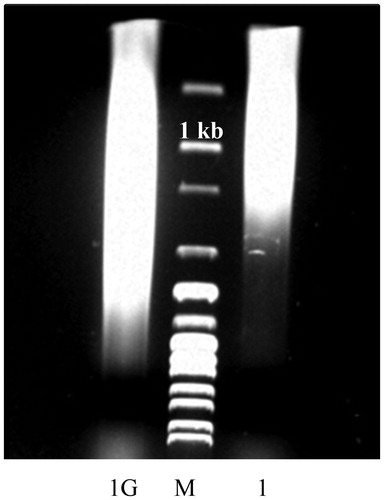
Until recently oxidation of DNA was thought to be the major endogenous cause of DNA damage and mutability [Citation89,Citation90] while the role of the Maillard reaction was unreasonably neglected [Citation30]. A role for the Maillard reaction in aging has been postulated by Cerami [Citation14] and Monnier [Citation15] in two independent studies soon after the realization that DNA is susceptible to glycation in vitro [Citation91]. Nevertheless, nearly two decades later John Baynes was still appealing ‘The Maillard hypothesis of aging: Time to focus on DNA’ [Citation92]. Glycation adducts in DNA had not been detected in vivo for a long time after the pioneer studies of Bucala et al. [Citation91], which could explain the delay in the research on the gerontological aspects of the Maillard reaction. It was, however, a matter of time and development of more sensitive analytical techniques to detect such products in DNA in vivo. We were first to show that the chromosomal DNA of E. coli accumulates Amadori products and AGEs under normal physiological conditions. Thereby, we were able to detect such products even by methods of moderate sensitivity like enzyme-linked immunosorbent assay, UV-vis, fluorescent, and colourimetric spectroscopy [Citation93]. In later studies, the research group of P. Thornalley, by applying high resolution mas-spectrometry, detected and quantitated several DNA-AGEs in tissues and body fluids of healthy human subjects and at elevated levels in diabetics as well [Citation29,Citation30,Citation94,Citation95]. We have recently observed that glycation of DNA with glucose 6-phosphate (G6P) results in significant gain in the molecular mass of the glycated DNA, which is indicative of DNA crosslinking (). Aldehydes [Citation96–98] and nitrous acid [Citation99] were shown to cause interstrand DNA crosslinking; however, DNA-AGEs crosslinks have not been characterized either qualitatively or quantitatively yet. To the best of our knowledge, also the question of whether DNA glycation correlates positively with age still awaits its answer and is a direction for future investigations.
Figure 5. Glycation-induced increase in the molecular mass of Salmon sperm DNA. DNA was sheared by sonication to an average size of 1 kb (1) and incubated for 2 weeks with 0.1 mol/L G6P at 37 °C under sterile conditions (1). Analysis was performed in 0.7% agarose gel. M, 1 kb DNA Ladder.
Glycation and conditional senescence of E. coli
Until the beginning of this century it was believed that bacteria such as E. coli, which divide symmetrically, do not exhibit an instant aging and are de facto immortal. What is reminiscent of ‘natural’ death of higher organisms seemed to occur in bacteria only under conditions of starvation-induced growth arrest and this kind of aging was referred to as ‘conditional senescence’ [Citation100–102]. Conditional senescence is caused by nutrient deprivation, overgrowth and accompanying quorum sensing [Citation103] during batch culturing of E. coli. Under such conditions, the life cycle of E. coli is composed of five phases [Citation104]. When transferred to a fresh nutrient-rich medium at a low density, E. coli experiences a latent period (lag phase) of several hours needed to adjust metabolically to the new environment before entering the active proliferative (exponential or logarithmic) phase. During this phase, E. coli divides rapidly (generation time ca. 20 min) over several hours. After exponential growth, batch cultured E. coli enters a stationary phase characterized by no obvious increase or decrease in the total number of cells. This phase lasts 2 to 3 days, during which most cells progressively loose replicative capacity but remain intact for extended periods of time [Citation105]. Thereafter follows the onset of the fourth, death phase, ending up with vigorous decay of the bacterial population. In this phase, up to 99% of the cells lyse and die, thereby releasing a large amount of cell debris.
In the last growth phase, called long-term stationary phase (LTSF), commencing in ca. 1-week-old batch cultures, some survivors of the death phase acquire the so-called ‘growth advantage in stationary phase’ (GASP) phenotype [Citation106,Citation107]. GASP means that if such cells are mixed with younger cells (e.g. from 1-day-old cultures), they will displace them after prolonged (7–10 days) co-culturing [Citation104]. It is remarkable that such cells may retain viability up to 10 years in liquid cultures without the addition of nutrients [Citation108]. High throughput proteomics and microarray studies as those published recently [Citation108,Citation109] are expected to provide insight into the heterogeneous genetic nature of this intriguing phenotype. Common feature of all GASP mutants is their increased ability to catabolize one or more amino acids as the sole energy source [Citation110–113]. It is still disputable whether E. coli conditional senescence is a programmed phenomenon, and in a detailed review on that topic, Nyström [Citation114] concluded that bacterial senescence is linked to stochastic deterioration rather than to programmed death pathways. He further suggested that self-inflicted oxidative damage may be a causal factor in age-related deterioration of both prokaryotes and higher organisms. The spontaneous (not genetically encoded) Maillard reaction may cause such a stochastic damage to life essential amines including proteins, DNA and amino-lipids and thus be an important player in the major events of E. coli senescence, which are mutability, death, and adaptation.
We have previously shown that the severity of the carbonyl stress in batch cultured E. coli, as mirrored by the glycation of the total protein and the chromosomal DNA, depends on the strain genotype, culture medium composition, and the growth phase [Citation93,Citation115–117]. AGEs accumulated predominantly in stationary phase cells and in rich versus minimal glucose supplemented medium. Subsequent independent studies have confirmed this observation [Citation118,Citation119]. The presence of reducing sugars such as glucose and fructose in the Luria-Bertani (LB) medium at concentrations above 0.2% caused reduced viability of cells in stationary phase and accelerated death in the death phase, which correlated positively with the accumulation of the advanced product of glycoxidation/lipoxidation CML in the total protein. In addition, the supplementation of the LB medium with 2 mmol/L of the highly potent glycating compound MG lowered dramatically (million fold) the viability of stationary cells [Citation118]. When LB was replaced with richer media containing higher concentrations of Yeast extract and Tryptone (Soytone), it was realized that the richer the medium, the more severe the death phase and the less the degree of recovery in the LTSP is [Citation119]. High levels of CML in the total protein accompanied the onset of the death phase for all media tested, and the severity of the death phase correlated positively with the spontaneous mutation rate to rifampicin resistance (Rifr) in late exponential cells. In these cells, the amount of AGEs-modified proteins depended also on the culture volume with bigger volumes promoting higher glycation levels [Citation120].
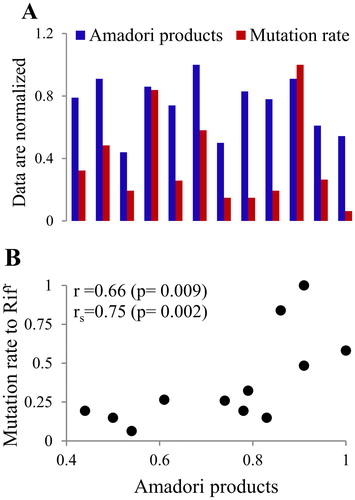
Kram and Finkel [Citation119,Citation120] demonstrated that the accumulation of AGEs in endogenous proteins is a hallmark of E. coli senescence. Importantly, there was a positive correlation between the level of protein-bound AGEs and the frequency of spontaneous mutations to Rifr in stationary cells. This correlation indirectly indicates that glycation of DNA is also a factor in bacterial senescence. By suppressing glycation adding various glycation inhibitors to the nutrient broth, we also found that there is a high positive correlation between the amount of Amadori products (AGEs-precursors) in the E. coli chromosomal DNA and the mutation frequency to Rifr phenotype in stationary phase E. coli cells [Citation117,Citation121] ().
Figure 6. The level of Amadori products in DNA and the spontaneous mutation rate to Rifr (A) in E. coli stationary phase cells exhibit high positive correlation (B). The Pearson (r) and the Spearman (rs) correlation coefficients are shown.
Glycation and replicative aging of E. coli
If there were no nutrient restriction (no starvation) and no invaders (phages, antibiotics) in the environment, would bacteria live (i.e. divide) forever? It is obvious that higher eukaryotes including humans do age and die despite having no food limitation and even more, life expectancy appears to be inversely related to the caloric intake [Citation122]. Does the same hold true for bacteria as well? One decade ago the answer to this question was rather negative. It was believed that bacteria that reproduce via binary fission do not exhibit instant (replicative) aging [Citation114,Citation123]. This belief was challanged by Stewart et al. [Citation17], who monitored the division of 94 single cells as they form colonies on a flat surface in real time via fluorescence microscopy. The authors observed that some cellular components with relatively long half-life and low diffusion rate accumulate predominantly at one of the two poles of the new-born E. coli cell, called the old pole. This is the pole inherited from the parent cell opposite to the division septum, which becomes the new pole. The old pole cells grew slowly and produced less offspring biomass, and deaths among them were more frequent as compared to the new pole cells. These observations imply that asymmetric division (in morphological terms) and juvenile phase are not mandatory for bacterial aging and that the basic principles of aging have to be searched at a molecular level.
Interesting conclusions came from the filamentation phenomenon observed during continuous fermentation of E. coli in the study of Wang et al. [Citation18], who used the so called ‘mother machine’ representing a high-throughput microfluidic liquid-culture devise ensuring long-term delivery of fresh medium to each cell. The filamentation phenotype (enlarged cells) results from cell growth that is not followed by division. While the filamentation rate of the new pole (daughter) cells remained constant over time (∼ 1 filamentation per 100 generations), there was more than 10 fold increase in the filamentation rate of the old pole (mother) cells with advancing replicative age. Because filamentation is a hallmark of the SOS response (global response to DNA damage), this means that the mother cells, inheriting the same old pole over many generations, more often switch on the SOS alarm due to DNA damage. This suggestion was confirmed by the increased mortality rate of an E. coli strain with blocked SOS response. Additionally, there was a positive correlation between the filamentation and death rates of the E. coli wild-type strain with a functional SOS response.
What is the cause of aging and death of individual cells in the expanding bacterial population? Wang et al. [Citation18] hypothesized that ‘the mother cell must inherit an unknown “factor” that serves as long-term memory from one generation to the next and causes filamentation’ and death. The unknown death-causing factor might be asymmetrically distributed aggregates in E. coli [Citation124,Citation125] and/or metabolic slowdown at the old pole cell wall [Citation126]. Reasonably, because of the high death rate, the authors excluded stochastic fluctuations in DNA damage as a cause of the observed mortality of the wild-type E. coli strain. However, accumulating (i.e. age-dependent) DNA damage should not be excluded as a probable death-causing factor. We are tempted to speculate that the robust E. coli growth under conditions of unlimited nutrient supply could be in part contributed by the diploid and tetraploid state of actively dividing E. coli (generation time ∼ 20 minutes). Under such conditions, bacterial cells are less susceptible to DNA damage, which therefore can accumulate and become evident in stationary phase cells because of their haploid state.
Does the Maillard reaction contribute to the replicative aging of E. coli? The pioneer studies of Stewart et al. [Citation17] and Wang et al. [Citation18] linked the aging mechanism of E. coli to the asymmetric distribution of some cellular components including protein aggregates. Next investigations indeed showed that heat shock as well as overexpression of recombinant proteins in E. coli result in the accumulation of protein aggregates in one of the cell poles [Citation124,Citation125]. Aggregation of native proteins in E. coli is promoted by stress conditions (oxidative stress, heat shock), as well as by errors during transcription and translation, resulting in aggregation even in the absence of an external assault [Citation127]. The contribution of glycation-mediated crosslinking to protein aggregation in E. coli is poorly studied. We have previously shown that while expressed in E. coli, human interferon-gamma (IFNγ) accumulates both Amadori products and AGEs, depending on the host strain and fermentation conditions [Citation16,Citation128,Citation129]. We also observed that some internal Arg and Lys residues, which are glycation targets, are involved in IFNγ crosslinking and covalent aggregation [Citation130,Citation131]. The protein was abundantly expressed (up to 50% of the total protein) and was sequestered by the bacterial cells in insoluble inclusion bodies with polar localization [Citation132] (). One can see that the E. coli inclusion bodies in are very similar to the protein aggregates accumulating in age-related human diseases such as the LB in PD (compare with ). Most studies specifically focus on the presence of protein aggregates in E. coli inclusion bodies; however, we have recently observed that IFNγ inclusion bodies can contain nucleic acids as well (plasmid DNA and rRNA) [Citation133–135]. As shown in , when glycated in vitro, DNA gains in molecular mass, which is indicative of DNA crosslinking. This result, together with our observation that E. coli DNA accumulates AGEs [Citation93], implies that DNA crosslinking and aggregation are likely to occur also in vivo. In addition, AGEs-crisscrosses between DNA, proteins and other primary amines might contribute to an even more complex structure of the E. coli inclusion bodies.
Figure 7. (A) Electron micrograph of E. coli cells overexpressing IFNγ; (B) Scheme of the coupled transcription and translation in E. coli as implicated in the formation of inclusion bodies and plasmid segregation.
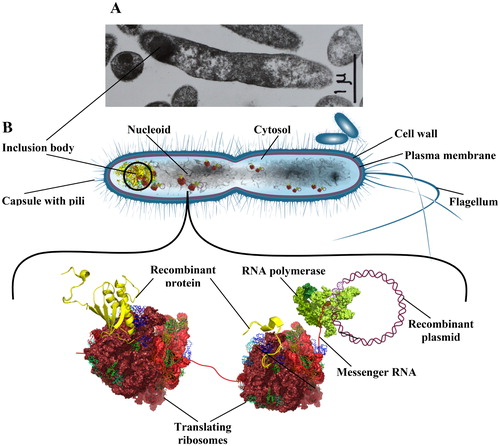
Yet another observation made by our group supports the E. coli aging mechanism proposed by Stewart et al. [Citation17] and Wang et al. [Citation18]. To express human IFNγ in E. coli, we cloned the IFNγ gene in a multicopy plasmid under a strong (phage) constitutive promoter. As the fermentation advanced, we observed a phenomenon called plasmid segregation. With time, some cells lost their plasmids, began to grow faster and displaced the plasmid-bearing cells, which resulted in reduced IFNγ yield. To explain this observation, we proposed a model based on the coupled transcription and translation in E. coli (). Because E. coli lacks a nuclear envelope, transcription takes place in the cytosol, where translation of the commencing mRNA starts before transcription is terminated. Assuming that IFNγ aggregation and inclusion bodies formation may occur while IFNγ is still being synthesized, our model predicts that the whole complex, including plasmid DNA, mRNA and the resulting IFNγ together with the supporting ribosomes (polysomes), might be tethered to the cell poles by the inclusion bodies and anchored into the cell membrane [Citation135]. This model explains why plasmid DNA and rRNA are present in the inclusion bodies and how plasmid segregation may occur. In fact, plasmid segregation seems to be tightly linked to the E. coli aging mechanism. The recombinant plasmids and the encoded protein impose a metabolic burden on E. coli cells and are therefore partitioned together with other surplus components in one of the two progeny cells, the mother cell, resulting in relieved growth of the daughter cell.
Conservative nature of the enzymatic defence against glycation
By applying various glycation inhibitors to the nutrient broth, we have shown that the protein/DNA glycation levels and the spontaneous mutation rate in E. coli are manageable [Citation16,Citation93,Citation117,Citation121,Citation131]. In the study of Kram and Finkel [Citation120], the anti-glycation agent carnosine exhibited pronounced inhibitory effect on the death phase, restoring the viable cell counts up to five orders of magnitude. Other compounds found to protect against high concentrations of glucose and MG were aminoguanidine and folic acid, while aspirin and grape seed extract abrogated the effect of MG but not that of glucose [Citation136]. In natural habitats, E. coli cannot rely on the delivery of such compounds and has elaborated diverse mechanisms for anti-glycation defence. These mechanisms operate at all steps of the Maillard reaction starting with detoxification of carbonyl compounds, going thorough degradation of Amadori products and ending up with excretion of AGEs.
One of the best-studied toxic carbonyl compounds in E. coli is MG [Citation137]. MG is produced in E. coli either spontaneously [Citation138] or enzymatically [Citation139,Citation140] and if not detoxified, its accumulation ultimately causes cell death [Citation141–144]. In all species, the main route for MG detoxification is the glyoxalase I/II system [Citation145,Citation146] and some data point to the presence of an additional glyoxalase III in E. coli [Citation147,Citation148]. Other enzymes involved in MG detoxification are aldose reductases [Citation149,Citation150] and enzymes converting MG to acetol encoded by the E. coli genes yghZ, yafB, yqhE, and yeaE [Citation151]. Two independent studies [Citation152,Citation153] revealed another group of enzymes in E. coli with glyoxalase/deglycase activity. These enzymes, HchA, YajL, YhbO, and ElbB, share similarity with the human DJ-1 superfamily of proteins. When overexpressed in E. coli, the bacterial homologs of human DJ-1, YajL, YhbO, and ElbB reduce the glyoxal (GO) dependent increase in intracellular AGEs [Citation153]. The apparent glyoxalase activity of YajL and YhbO seems to reflect their deglycase activity as far as these two enzymes are capable of repairing GO- and MG-glycated proteins [Citation152]. The sequence similarity of the four E. coli proteins with human DJ-1, known to be associated with PD, and their expression primarily during E. coli stationary phase [Citation153], point to the existence of common mechanisms for defence against carbonyl stress and senescence from bacteria to humans.
The removal of fructosamines (Amadori products) formed by glucose on human haemoglobin is carried out by the enzyme fructosamine 3-kinase, acting as a deglycase [Citation154,Citation155]. This enzyme phosphorylates the fructose residue at the third carbon atom, thus promoting the spontaneous breakdown of the bond between the protein and the reducing sugar. Fructosamine 3-kinase analogue was not found in E. coli [Citation156], where the decomposition of Amadori products was shown to result from the cooperative action of two enzymes, a kinase (FrlD) and a deglycase (FrlB) [Citation157]. These enzymes utilize as a substrate G6P-derived Amadori products bound to the ε-NH2 group of free lysine, not embedded in proteins. Therefore, they do not repair glycated proteins but rather sustain bacterial growth on fructoselysine, released during digestion of glycated proteins in the human intestine [Citation157]. Another E. coli enzyme, the Gcp glycopeptidase, has been suggested to directly repair Amadori products-modified proteins (AMPs), because: (i) Gcp binds glycated proteins, (ii) its depletion results in accumulation of AMPs and (iii) the severe phenotype of Gcp depletion can be relieved under conditions of low intracellular glycation [Citation158]. Noteworthily, Gcp is a conserved enzyme encoded by nearly every sequenced genome in all three domains of life, suggesting a universal involvement of Gcp in cellular aging. By analogy to the excretion of AGEs in the urine of mammals, E. coli seems to have developed mechanisms for active excretion of AGEs outside cells in the form of free AGEs (bound to amino acids) and peptide-AGEs. It has been shown that low-molecular-weight AGEs originate from high-molecular-weight AGEs by proteolytic degradation, not carried out by the major ATP-dependent proteases but by an alternative metal-dependent proteolysis [Citation159,Citation160].
Repair of glycation damaged DNA
The mechanisms for repair of glycation damaged DNA are poorly understood. First studies provided data about repair of the product of the advanced DNA glycation N2-(1-carboxyethyl)-2′-deoxyguanosine (CEdG) [Citation161]. This is one of the major stable DNA-AGEs, resulting from the interaction of DNA with MG. CEdG has been detected in vitro [Citation162] as well as in mononuclear leukocytes’ DNA, plasma and urine of healthy human subjects [Citation29,Citation163] and at increased levels in body fluids [Citation95], kidneys and aortas [Citation164] of diabetic patients. This DNA adduct was shown to be weakly mutagenic to E. coli, and DinB (pol IV), which belongs to the Y-family of translesion DNA polymerases, is the major enzyme responsible for bypassing the lesion in vivo. Steady-state kinetic measurements revealed that nucleotide insertion, catalyzed by E. coli pol IV or its human counterpart (polymerase κ), opposite the CEdG lesion is both accurate and efficient. Pol IV is known to be constitutively expressed in E. coli and highly up-regulated together with pol V upon SOS induction [Citation165]. On the other hand, the SOS response is involved in the regulation of the adaptive (stationary-phase) mutagenesis in E. coli upon starvation [Citation166]. Taken together these data imply that translesion DNA synthesis might be implicated in both accurate and error-prone repair of CEdG lesions in DNA of stationary phase E. coli cells. In a later study, Tamae et al. [Citation167] provided data about the involvement of NER in the repair of the same lesion (CEdG) in human fibroblasts in vitro, which suggests that CEdG might be repaired by alternative pathways. Interestingly, at high CEdG levels, the authors found a significant increase in the frequency of AT → GC transitions in NER-competent cells compared to NER-deficient cells. This observation led them to suppose that a NER-dependent mutagenic process is associated with the repair of heavily damaged DNA, possibly through the error-prone Y-family of DNA polymerases. Finally, it is worth noting that the Y-family of DNA polymerases including E. coli pol IV and V and human pol κ and η are highly conserved among all domains of life [Citation161,Citation168].
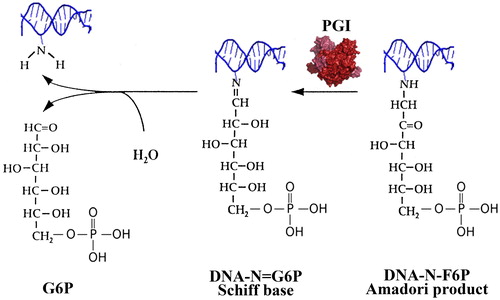
The early products of DNA glycation (Schiff’s bases and Amadori products) are precursors for the formation of DNA-AGEs. It is therefore reasonable to assume that there must be mechanisms for repair of early glycation adducts in DNA in order to preclude involvement of DNA in more dramatic chemical rearrangements such as interstrand crosslinks and double-strand breaks. In the same year, when Yuan et al. [Citation161] reported on the repair of CEdG, we postulated the existence of DNA amdoriase(s) in E. coli [Citation117]. At that time we conducted experiments to test the E. coli FrlB amadoriase [Citation157] for deglycation activity with substrate G6P-modified DNA (DNA-N-fructosamine-6-phosphate, DNA-N-F6P). We did not observe DNA deglycation activity with FrlB but E. coli lysates proved to be DNA-N-F6P deglycation competent. In subsequent studies we identified the glycolytic enzyme phosphoglucose isomerase (PGI) as a carrier of the observed DNA-N-F6P amadoriase activity. In a recently published work, we demonstrated that PGI catalyses the release of G6P from G6P-modified single stranded DNA oligonucleotide and from its hybrid duplex with a complementary peptide nucleic acid [Citation169]. Also, we have observed an increased spontaneous mutation rate to Rifr of an E. coli strain with a deleted pgi gene, which points to the possible DNA repair activity of PGI in vivo.
In early glycolysis PGI does catalyze the isomerization of G6P to fructose 6-phosphate (F6P). This reaction is reversible in vitro and particularly for yeasts and E. coli PGI it has been shown that the reaction is slightly favourable in the reverse direction, that is from F6P to G6P (Keq ∼ 0.3) [Citation170,Citation171]. Therefore, we suppose that PGI recognizes F6P bound to DNA in the form of an Amadori product and serves to reverse the poorly reversible step from Amadori produts (DNA-N-F6P) to Schiff’s bases (DNA-N = G6P), which then, because of their reversibility, can spontaneously hydrolyse to free (repaired) DNA and G6P (). The deglycation mechanism that we propose here is supported by the fact that the FrlB amadoriase, which recognizes the same Amadori product, albeit linked to the lysine ε-amino group, is homologous to the isomerase domain of glucosamine-6-phosphate synthase [Citation157]. Applying bioinformatics approaches, we found that both enzymes, FrlB and PGI, belong to the sugar isomerase superfamily (SIS) of enzymes and share two common SIS domains, one for binding the phosphorylated sugar (SIS1) and the other one for accomplishment of the isomerization reaction (SIS2). Therefore, it appears that the deglycation mechanism of PGI and FrlB is tightly linked to their intrinsic isomerase activity. PGI is a highly conserved enzyme from Archaea to humans with respect to both 3D-structure and catalytic (isomerization) mechanism [Citation172,Citation173]. Therefore, like many other mechanisms discussed in this review, the anti-glycation defence against G6P-damaged DNA by PGI also appears to be highly conserved among species.
Figure 8. Proposed DNA-N-F6P deglycation mechanism of PGI: Reversal of the early step of the Maillard reaction.
Richarme et al. [Citation174] proposed to reconsider the semantics of the terms ‘early’ and ‘late’ glycation adducts with respect to glycation by dicarbonyl compounds. In fact, the formation of CEdG from 2′-deoxyguanosine (dG) and MG is preceded by discrete chemical steps: (i) addition reaction resulting in the formation of an aminocarbinol (N2-(1-hydroxy-2-oxopropyl)-2′-deoxyguanosine), (ii) structural isomerization of the aminocarbinol to two imidazopurinones 3-(2′-deoxyribosyl)-6,7-dihydro-6,7-dihydroxy-6/7-methylimidazo-[2,3-b]purine-9(8)one (MGdG) and (iii) slow transformation of MGdG to two stereoisomers N2-(1,R/S-carboxyethyl)-2′-deoxyguanosine (CEdG). Analogous products are formed by glycation of dG with GO – the aminocarbinol (N2-(1-hydroxy-2-oxoethyl)-2′-deoxyguanosine, the imidazopurinone 3-(2′-deoxyribosyl)-6,7-dihydro-6,7-dihydroxyimidazo-[2,3-b]purine-9(8)one (GdG) and the product of the advanced glycation N2-(1-carboxymethyl)-2′-deoxyguanosine (CMdG) [Citation29,Citation95]. Aminocarbionls appear over minutes and hours after mixing nucleotides with GO or MG, whereas M/GdG and CM/EdG arise after hours and days of incubation. Also, the formation of all these products does not go through Schiff’s bases and Amadori products as in the classical Maillard reaction initiated by aldoses (e.g. glucose). Therefore, in the pathway of DNA glycation by GO and MG, aminocarbinols should be considered ‘early’, whereas M/GdG and CM/EdG ‘late’ glycation products. After these terminological details, we can specify that Richarme et al. [Citation175] reported on a deglycation activity evolved to repair early glycation adducts in DNA.
The human DJ-1 deglycase and its bacterial homologs, shown by Richarme et al. [Citation174] to repair GO- and MG-glycated amino acids, was reported by the same authors to act also as a nucleotide sanitizer by repairing GTP, dGTP, GDP, and GMP modified by aminocarbinols before their conversion into AGEs. The E. coli homologs of DJ-1, the proteins HchA (Hsp31) and YhbO, efficiently deglycated GTP, whereas YajL deglycated these compounds with lower efficiency. In in vitro studies, DJ-1 exhibited broad substrate specificity including GO/MG-modified nucleotides, single/double stranded DNA and RNA as well. The in vivo significance of these observations, however, is questioned by the studies of Lo et al. [Citation176] and Rabbani and Thornalley [Citation177] because of the low catalytic constant (Kcat = 0.25 s−1) and the spontaneous reversal of the DJ-1 catalyzed reaction. In fact, in the study of Richarme et al. [Citation175] the in vivo effect of DJ-1 was demonstrated indirectly, by the increased DNA glycation levels and the enhanced mutagenesis in E. coli and Hela cells with knock-out or silenced, respectively, deglycase genes. Intriguingly, the E. coli mutant strains demonstrated an impressive mutator phenotype, which hyperbolically illustrates what would happen in case of insufficiency or failure of the DNA repair systems with advancing age.
Conclusions
The answer to the question that we asked in the title of this review, ‘Maillard reaction and aging: can bacteria shed light on the link?’ is definitely affirmative. Current high-throughput live cell imaging helped debunk the myth of bacterial immortality and provided evidence that non-stochastic deaths among proliferating E. coli cells are the outcome of an on-going aging process. The signs of aging were recognized in the functional asymmetry between the two E. coli progeny cells. Accumulating damage in the old pole mother cells in the form of protein aggregates is very reminiscent of the protein aggregation observed in human age-related diseases such as PD and AD. It appears that through promoting crosslinking and aggregation of proteins, DNA and other amines, the Maillard reaction (glycation) contributes to molecular deterioration during aging of both bacterial and human cells. Finally, the pathways for defence against glycation are highly conserved among species, which gives us hope that E. coli may provide an excellent platform for deciphering the intimate mechanisms of aging.
Disclosure of interest
The authors report no conflict of interest.
Additional information
Funding
Related Research Data
References
- Medawar PB. An unsolved problem of biology. 1st ed. London: Lewis; 1952.
- Williams GC. Pleiotropy, natural selection and the evolution of senescence. Evolution. 1957;11:398–411.
- Kirkwood TB. Evolution of ageing. Nature. 1977;270:301–304.
- Darwin CR. On the origin of species by means of natural selection, or the preservation of favoured races in the struggle for life. 1st ed. London: John Murray; 1859.
- Gallant J, Palmer L. Error propagation in viable cells. Mech Ageing Dev. 1979;10:27–38.
- Barton A. Some aspects of cell division in Saccharomyces cerevisiae. J Gen Microbiol. 1950;4:84–86.
- Mortimer RK, Johnston JR. Life span of individual yeast cells. Nature. 1959;183:1751–1752.
- Nyström T, Liu B. The mystery of aging and rejuvenation - a budding topic. Curr Opin Microbiol. 2014;18:61–67.
- Higuchi-Sanabria R, Pernice WM, Vevea JD. Role of asymmetric cell division in lifespan control in Saccharomyces cerevisiae. FEMS Yeast Res. 2014;14:1133–1146.
- Hill SM, Hao X, Grönvall J, et al. Asymmetric inheritance of aggregated proteins and age reset in yeast are regulated by Vac17-dependent vacuolar functions. Cell Rep. 2016;16:826–838.
- Szilard L. On the nature of the aging process. Proc Natl Acad Sci USA. 1959;45:30–45.
- Alexander P. The role of DNA lesions in the processes leading to aging in mice. Symp Soc Exp Biol. 1967;21:29–50.
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300.
- Cerami A. Aging of proteins and nucleic acids: what is the role of glucose?. Trends Biochem Sci. 1986;11:311–314.
- Monnier VM. Toward a Maillard reaction theory of aging. Prog Clin Biol Res. 1989;304:1–22.
- Mironova R, Niwa T, Hayashi H, et al. Evidence for non-enzymatic glycosylation in Escherichia coli. Mol Microbiol. 2001;39:1061–1068.
- Stewart EJ, Madden R, Paul G, et al. Aging and death in an organism that reproduces by morphologically symmetric division. PLoS Biol. 2005;3:e45.
- Wang P, Robert L, Pelletier J, et al. Robust growth of Escherichia coli. Curr Biol. 2010;20:1099–1103.
- Maillard LC. Action des acides amines sur les sucres: formation des melanoidines par voie methodique [Action of amino acids on sugars: method for melanoidines formation]. C R Acad Sci. 1912;154:66–68.
- Hodge JE. The Amadori rearrangement. Adv Carbohydr Chem. 1955;10:169–205.
- Nakayama T, Hayase F, Kato H. Formation of Nε-(2-formyl-5-hydroxy-methyl-pyrrol-1-yl)-L-norleucine in the Maillard reaction between D-glucose and L-lysine. Agric Biol Chem. 1980;44:1201–1202.
- Pongor S, Ulrich PC, Bencsath FA, et al. A. Aging of proteins: isolation and identification of a fluorescent chromophore from the reaction of polypeptides with glucose. Proc Natl Acad Sci USA. 1984;81:2684–2688.
- Ahmed MU, Thorpe SR, Baynes JW. Identification of N epsilon-carboxymethyllysine as a degradation product of fructoselysine in glycated protein. J Biol Chem. 1986;261:4889–4894.
- Sell DR, Monnier VM. Structure elucidation of a senescence cross-link from human extracellular matrix. Implication of pentoses in the aging process. J Biol Chem. 1989;264:21597–21602.
- Ahmed N, Thornalley PJ. Quantitative screening of protein biomarkers of early glycation, advanced glycation, oxidation and nitrosation in cellular and extracellular proteins by tandem mass spectrometry multiple reaction monitoring. Biochem Soc Trans. 2003;31:1417–1422.
- Thornalley PJ, Battah S, Ahmed N, et al. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem J. 2003;375:581–592.
- Thorpe SR, Baynes JW. Maillard reaction products in tissue proteins: new products and new perspectives. Amino Acids. 2003;25:275–281.
- Lapolla A, Molin L, Traldi P. Protein glycation in diabetes as determined by mass spectrometry. Int J Endocrinol. 2013;2013:412103.
- Thornalley PJ, Waris S, Fleming T, et al. Imidazopurinones are markers of physiological genomic damage linked to DNA instability and glyoxalase 1-associated tumour multidrug resistance. Nucleic Acids Res. 2010;38:5432–5442.
- Rabbani N, Thornalley PJ. Dicarbonyl proteome and genome damage in metabolic and vascular disease. Biochem Soc Trans. 2014;42:425–432.
- Rahbar S. An abnormal hemoglobin in red cells of diabetics. Clin Chim Acta. 1968;22:296–298.
- Brownlee M. Advanced protein glycosylation in diabetes and aging. Annu Rev Med. 1995;46:223–234.
- Thorpe SR, Baynes JW. Role of the Maillard reaction in diabetes mellitus and diseases of aging. Drugs Aging. 1996;9:69–77.
- Ulrich P, Cerami A. Protein glycation, diabetes, and aging. Recent Prog Horm Res. 2001;56:1–21.
- Nass N, Bartling B, Navarrete Santos A, et al. Advanced glycation end products, diabetes and ageing. Z Gerontol Geriatr. 2007;40:349–356.
- Hellwig M, Henle T. Baking, ageing, diabetes: a short history of the Maillard reaction. Angew Chem Int Ed Engl. 2014;53:10316–10329.
- Sadowska-Bartosz I, Bartosz G. Effect of glycation inhibitors on aging and age-related diseases. Mech Ageing Dev. 2016;160:1–18.
- Semba RD, Nicklett EJ, Ferrucci L. Does accumulation of advanced glycation end products contribute to the aging phenotype?. J Gerontol A Biol Sci Med Sci. 2010;65:963–975.
- Bjorksten J. A common molecular basis for the aging syndrome. J Am Geriatr Soc. 1958;8:740–748.
- Bjorksten J, Tenhu H. The crosslinking theory of aging-added evidence. Exp Gerontol. 1990;25:91–95.
- Cárdenas-León M, Díaz-Díaz E, Argüelles-Medina R, et al. Glycation and protein crosslinking in the diabetes and ageing pathogenesis. Rev Invest Clin. 2009;61:505–520.
- Monnier VM, Mustata GT, Biemel KL, et al. Cross-linking of the extracellular matrix by the Maillard reaction in aging and diabetes: an update on ‘puzzle nearing resolution’. Ann N Y Acad Sci. 2005;1043:533–544.
- Beisswenger PJ, Howell S, Mackenzie T, et al. Two fluorescent wavelengths, 440(ex)/520(em) nm and 370(ex)/440(em) nm, reflect advanced glycation and oxidation end products in human skin without diabetes. Diabetes Technol Ther. 2012;14:285–292.
- Couppé C, Hansen P, Kongsgaard M, et al. Mechanical properties and collagen cross-linking of the patellar tendon in old and young men. J Appl Physiol. 2009;107:880–886.
- Snedeker JG, Gautieri A. The role of collagen crosslinks in ageing and diabetes - the good, the bad, and the ugly. Muscles Ligaments Tendons J. 2014;4:303–308.
- Verzijl N, DeGroot J, Thorpe SR, et al. Effect of collagen turnover on the accumulation of advanced glycation end products. J Biol Chem. 2000;275:39027–39031.
- Verzijl N, DeGroot J, Ben ZC, et al. Crosslinking by advanced glycation end products increases the stiffness of the collagen network in human articular cartilage: a possible mechanism through which age is a risk factor for osteoarthritis. Arthritis Rheum. 2002;46:114–123.
- Saito M, Marumo K, Fujii K, et al. Single-column high-performance liquid chromatographic-fluorescence detection of immature, mature, and senescent cross-links of collagen. Anal Biochem. 1997;253:26–32.
- Saito M. Age-related changes in biochemical characteristics of collagen from human weight-bearing and non-weight bearing bone. Tokyo Jikeikai Med J. 1999;114:327–337.
- Wang X, Shen X, Li X, et al. Age-related changes in the collagen network and toughness of bone. Bone. 2002;31:1–7. Erratum in: Bone. 2003;32(1):107.
- Saito M, Fujii K, Mori Y, et al. Role of collagen enzymatic and glycation induced cross-links as a determinant of bone quality in spontaneously diabetic WBN/Kob rats. Osteoporos Int. 2006;17:1514–1523.
- Sady C, Khosrof S, Nagaraj R. Advanced Maillard reaction and crosslinking of corneal collagen in diabetes. Biochem Biophys Res Commun. 1995;214:793–797.
- Aronson D. Cross-linking of glycated collagen in the pathogenesis of arterial and myocardial stiffening of aging and diabetes. J Hypertens. 2003;21:3–12.
- Sims TJ, Rasmussen LM, Oxlund H, et al. The role of glycation cross-links in diabetic vascular stiffening. Diabetologia. 1996;39:946–951.
- Salahuddin P, Rabbani G, Khan RH. The role of advanced glycation end products in various types of neurodegenerative disease: a therapeutic approach. Cell Mol Biol Lett. 2014;19:407–437.
- Muronetz VI, Melnikova AK, Seferbekova ZN, et al. Glycolysis, and neurodegenerative diseases: is there any connection?. Biochemistry (Moscow). 2017;82:874–886.
- Thomas B, Beal MF. Molecular insights into Parkinson's disease. F1000 Med Rep. 2011;3:7.
- WIKIMEDIA COMMONS [Internet]: Marvin 101 - Own work, CC BY-SA3.0. Available from: https://commons.wikimedia.org/w/index.php?curid=7533521
- WIKIMEDIA COMMONS [Internet]: Nephron - Own work, CC BY-SA 3.0. Available from: https://commons.wikimedia.org/w/index.php?curid=12274694
- Plotegher N, Bubacco L. Lysines, Achilles' heel in alpha-synuclein conversion to a deadly neuronal endotoxin. Ageing Res Rev. 2016;26:62–71.
- Haines DD, Trushin MV, Rose S, et al. Parkinson's disease: Alpha synuclein, heme oxygenase and biotherapeutic countermeasures. Curr Pharm Des. 2018;24:2317–2321.
- Dalfó E, Portero-Otín M, Ayala V, et al. Evidence of oxidative stress in the neocortex in incidental Lewy body disease. J Neuropathol Exp Neurol. 2005;64:816–830.
- Kurz A, Rabbani N, Walter M, et al. Alpha-synuclein deficiency leads to increased glyoxalase I expression and glycation stress. Cell Mol Life Sci. 2011;68:721–733.
- Münch G, Lüth HJ, Wong A, et al. Crosslinking of alpha-synuclein by advanced glycation endproducts - an early pathophysiological step in Lewy body formation?. J Chem Neuroanat. 2000;20:253–257.
- Choi YG, Lim S. N(ɛ)-(carboxymethyl)lysine linkage to α-synuclein and involvement of advanced glycation end products in α-synuclein deposits in an MPTP-intoxicated mouse model. Biochimie. 2010;92:1379–1386.
- Lee D, Park CW, Paik SR, et al. The modification of alpha-synuclein by dicarbonyl compounds inhibits its fibril-forming process. Biochim Biophys Acta. 2009;1794:421–430.
- Kö Nig A, Vicente Miranda H, Outeiro TF. Alpha-synuclein glycation and the action of anti-diabetic agents in Parkinson's disease. J Parkinsons Dis. 2018;8:33–43.
- Wong A, Lüth HJ, Deuther-Conrad W, et al. Advanced glycation endproducts co-localize with inducible nitric oxide synthase in Alzheimer's disease. Brain Res. 2001;920:32–40.
- Reddy VP, Obrenovich ME, Atwood CS, et al. Involvement of Maillard reactions in Alzheimer disease. Neurotox Res. 2002;4:191–209.
- Chen K, Maley J, Yu PH. Potential inplications of endogenous aldehydes in beta-amyloid misfolding, oligomerization and fibrillogenesis. J Neurochem. 2006;99:1413–1424.
- Münch G, Westcott B, Menini T, et al. Advanced glycation endproducts and their pathogenic roles in neurological disorders. Amino Acids. 2012;42:1221–1236.
- Münch G, Mayer S, Michaelis J, et al. Influence of advanced glycation end-products and AGE-inhibitors on nucleation-dependent polymerization of beta-amyloid peptide. Biochim Biophys Acta. 1997;1360:17–29.
- Freitas AA, de Magalhães JP. A review and appraisal of the DNA damage theory of ageing. Mutat Res. 2011;728:12–22.
- Liu B, Wang J, Chan KM, et al. Genomic instability in laminopathy-based premature aging. Nat Med. 2005;11:780–785.
- Mostoslavsky R, Chua KF, Lombard DB, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329.
- Holcomb VB, Vogel H, Hasty P. Deletion of Ku80 causes early aging independent of chronic inflammation and Rag-1-induced DSBs. Mech Ageing Dev. 2007;128:601–608.
- Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475–1485. Erratum in: N Engl J Med. 2009;361(19):1914.
- Iyama T, Wilson DM. 3rd. Elements that regulate the DNA damage response of proteins defective in Cockayne syndrome. J Mol Biol. 2016;428:62–78.
- Kanfi Y, Naiman S, Amir G, et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483:218–221.
- De Luca G, Ventura I, Sanghez V, et al. Prolonged lifespan with enhanced exploratory behavior in mice overexpressing the oxidized nucleoside triphosphatase hMTH1. Aging Cell. 2013;12:695–705.
- Cho M, Suh Y. Genome maintenance and human longevity. Curr Opin Genet Dev. 2014;26:105–115.
- Dominick G, Bowman J, Li X, et al. mTOR regulates the expression of DNA damage response enzymes in long-lived Snell dwarf, GHRKO, and PAPPA-KO mice. Aging Cell. 2017;16:52–60.
- White MC, Holman DM, Boehm JE, et al. Age and cancer risk: a potentially modifiable relationship. Am J Prev Med. 2014;46:S7–S15.
- Smetana K, Jr, Lacina L, Szabo P, et al. Ageing as an important risk factor for cancer. Anticancer Res. 2016;36:5009–5017.
- Aunan JR, Cho WC, Søreide K. The biology of aging and cancer: a brief overview of shared and divergent molecular hallmarks. Aging Dis. 2017;8:628–642.
- Acharya PV. The isolation and partial characterization of age-correlated oligo-deoxyribo-ribonucleotides with covalently linked aspartyl-glutamyl polypeptides. Johns Hopkins Med J Suppl. 1972;(1:):254–260.
- Ermolaeva MA, Schumacher B. Systemic DNA damage responses: organismal adaptations to genome instability. Trends Genet. 2014;30:95–102.
- Ribezzo F, Shiloh Y, Schumacher B. Systemic DNA damage responses in aging and diseases. Semin Cancer Biol. 2016;37-38:26–35.
- Ames BN, Gold LS. Endogenous mutagens and the causes of aging and cancer. Mutat Res. 1991;250:3–16.
- Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci USA. 1993;90:7915–7922.
- Bucala R, Model P, Cerami A. Modification of DNA by reducing sugars: a possible mechanism for nucleic acid aging and age-related dysfunction in gene expression. Proc Natl Acad Sci USA. 1984;81:105–109.
- Baynes JW. The Maillard hypothesis on aging: time to focus on DNA. Ann N Y Acad Sci. 2002;959:360–367.
- Mironova R, Niwa T, Handzhiyski Y, et al. Evidence for non-enzymatic glycosylation of Escherichia coli chromosomal DNA. Mol Microbiol. 2005;55:1801–1811.
- Rabbani N, Shaheen F, Anwar A, et al. Assay of methylglyoxal-derived protein and nucleotide AGEs. Biochem Soc Trans. 2014;42:511–517.
- Waris S, Winklhofer-Roob BM, Roob JM, et al. Increased DNA dicarbonyl glycation and oxidation markers in patients with type 2 diabetes and link to diabetic nephropathy. J Diabetes Res. 2015;2015:1.
- Stone MP, Cho YJ, Huang H, et al. Interstrand DNA cross-links induced by alpha,beta-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc Chem Res. 2008;41:793–804.
- Niedernhofer LJ, Daniels JS, Rouzer CA, et al. Malondialdehyde, a product of lipid peroxidation, is mutagenic in human cells. J Biol Chem. 2003;278:31426–31433.
- Dooley PA, Zhang M, Korbel GA, et al. NMR determination of the conformation of a trimethylene interstrand cross-link in an oligodeoxynucleotide duplex containing a 5'-d(GpC) motif. J Am Chem Soc. 2003;125:62–72.
- Edfeldt NB, Harwood EA, Sigurdsson ST, et al. Solution structure of a nitrous acid induced DNA interstrand cross-link. Nucleic Acids Res. 2004;32:2785–2794.
- Burleigh IG, Dawes EA. Studies on the endogenous metabolism and senescence of starved Sarcina lutea. Biochem J. 1967;102:236–250.
- Nyström T. Starvation, cessation of growth and bacterial aging. Curr Opin Microbiol. 1999;2:214–219.
- Nyström T. Not quite dead enough: on bacterial life, culturability, senescence, and death. Arch Microbiol. 2001;176:159–164.
- Miller MB, Bassler BL. Quorum sensing in bacteria. Annu Rev Microbiol. 2001;55:165–199.
- Finkel SE. Long-term survival during stationary phase: evolution and the GASP phenotype. Nat Rev Microbiol. 2006;4:113–120.
- Ericsson M, Hanstorp D, Hagberg P, et al. Sorting out bacterial viability with optical tweezers. J Bacteriol. 2000;182:5551–5555.
- Zambrano MM, Kolter R. GASPing for life in stationary phase. Cell. 1996;86:181–184.
- Vulic M, Kolter R. Evolutionary cheating in Escherichia coli stationary phase cultures. Genetics. 2001;158:519–526.
- Gagliardi A, Lamboglia E, Bianchi L, et al. Proteomics analysis of a long-term survival strain of Escherichia coli K-12 exhibiting a growth advantage in stationary-phase (GASP) phenotype. Proteomics. 2016;16:963–972.
- Arunasri K, Adil M, Khan PA, et al. Global gene expression analysis of long-term stationary phase effects in E. coli K12 MG1655. PLoS One. 2014;9:e96701.
- Zinser ER, Kolter R. Escherichia coli evolution during stationary phase. Res Microbiol. 2004;155:328–336.
- Zinser ER, Kolter R. Mutations enhancing amino acid catabolism confer a growth advantage in stationary phase. J Bacteriol. 1999;181:5800–5807.
- Zinser ER, Kolter R. Prolonged stationary-phase incubation selects for lrp mutations in Escherichia coli K-12. J Bacteriol. 2000;182:4361–4365.
- Zinser ER, Schneider D, Blot M, et al. Bacterial evolution through the selective loss of beneficial Genes. Trade-offs in expression involving two loci. Genetics. 2003;164:1271–1277.
- Nyström T. Conditional senescence in bacteria: death of the immortals. Mol Microbiol. 2003;48:17–23.
- Dimitrova R, Mironova R, Ivanov I. Glycation of proteins in Escherichia coli: Interference of strain diversity and growth conditions with glycation. C R Bulg Acad Sci. 2004;57:71–75.
- Dimitrova R, Mironova R, Ivanov I. Glycation of proteins in Escherichia coli: Effect of nutrient broth ingredients on glycation. Biotechnol Biotechnol Equip. 2004;18:99–103.
- Mironova R, Handzhiyski Y, T N, et al. Maillard reaction and spontaneous mutagenesis in Escherichia coli. In: Knutsen DW, Bruns SS, editors. Bacterial DNA, DNA polymerase and DNA helicases. New York (NY): Nova Science Publishers; 2008. p. 51–89.
- Pepper ED, Farrell MJ, Nord G, et al. Antiglycation effects of carnosine and other compounds on the long-term survival of Escherichia coli. Appl Environ Microbiol. 2010;76:7925–7930.
- Kram KE, Finkel SE. Rich medium composition affects Escherichia coli survival, glycation, and mutation frequency during long-term batch culture. Appl Environ Microbiol. 2015;81:4442–4450.
- Kram KE, Finkel SE. Culture volume and vessel affect long-term survival, mutation frequency, and oxidative stress of Escherichia coli. Appl Environ Microbiol. 2014;80:1732–1738.
- Handzhiyski Y, Mironova R, Ivanov I. Effect of acetyl salicyilic acid on glycation and mutability of Escherichia coli chromosomal DNA. Biotechnol Biotechnol Equip. 2009;23:1079–1083.
- Colman RJ, Beasley TM, Kemnitz JW, et al. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat Commun. 2014;5:3557.
- Nyström T. Aging in bacteria. Curr Opin Microbiol. 2002;5:596–601.
- Lindner AB, Madden R, Demarez A, et al. Asymmetric segregation of protein aggregates is associated with cellular aging and rejuvenation. Proc Natl Acad Sci USA. 2008;105:3076–3081.
- Winkler J, Seybert A, König L, et al. Quantitative and spatio-temporal features of protein aggregation in Escherichia coli and consequences on protein quality control and cellular ageing. EMBO J. 2010;29:910–923.
- den Blaauwen T, de Pedro MA, Nguyen-Distèche M, et al. Morphogenesis of rod-shaped sacculi. FEMS Microbiol Rev. 2008;32:321–344.
- Mogk A, Deuerling E, Vorderwülbecke S, et al. Small heat shock proteins, ClpB and the DnaK system form a functional triade in reversing protein aggregation. Mol Microbiol. 2003;50:585–595.
- Tsekovska RG, Boyanova МS, Mironova RS, et al. Effect of arginine on glycation and stability of recombinant human interferon-gamma. Biotechnol Biotechnol Equip. 2009;23:1063–1067.
- Tsekovska RG, Boyanova MS, Mironova RS, et al. Impact of glycation inhibitors on the biologic activity of recombinant human interferon-gamma. Biotechnol Biotechnol Equip. 2012;26:170–174. Sе:
- Mironova R, Niwa T, Dimitrova R, et al. Glycation and post-translational processing of human interferon-gamma expressed in Escherichia coli. J Biol Chem. 2003;278:51068–51074.
- Boyanova M, Mironova R, Niwa T, et al. Post-translational processing of human interferon-gamma and approaches for its prevention. In: Georgiev VS, Western KA, McGovan, JJ, editors. Infectious disease. Totowa (NJ): Humana Press; 2008. p. 365–373.
- Tsanev R, Ivanov I. Immune interferon. 1st ed. New York (NY): CRC Press; 2002.
- Ahmed IH, Krachmarova E, Nacheva G, et al. Nucleic acids in inclusion bodies obtained from E. coli cells expressing human interferon-gamma. Poster session presented at: The 8th Poster Session of the University of Chemical Technology and Metallurgy; May 18; Sofia, Bulgaria, 2011.
- Krachmarova E, Nacheva G, Ivanov I. Inclusion bodies obtained from E. coli cells expressing human interferon-gamma contain nucleic acids. In: Abstract book of the Anniversary Molecular Biology Conference ‘50 Year Roumen Tsanev Institute of Molecular Biology’, Sofia, Bulgaria; 2011 Oct 6–7, p. 90.
- Popov M, Petrov S, Nacheva G, et al. Effects of a recombinant gene expression on ColE1-like plasmid segregation in Escherichia coli. BMC Biotechnol. 2011;11:18.
- Pepper ED. Mechanisms of long-term survival in Escherichia coli (dissertation). Los Angeles (CA): University of Southern California; 2007.
- Kosmachevskaya OV, Shumaev KB, Topunov AF. Carbonyl stress in bacteria: Causes and consequences. Biochemistry Mosc. 2015;80:1655–1671.
- Ahmed MU, Brinkmann Frye E, Degenhardt TP, et al. N-epsilon-(carboxyethyl)lysine, a product of the chemical modification of proteins by methylglyoxal, increases with age in human lens proteins. Biochem J. 1997;324: 565–570.
- Hopper DJ, Cooper RA. The purification and properties of Escherichia coli methylglyoxal synthase. Biochem J. 1972;128:321–329.
- Tötemeyer S, Booth NA, Nichols WW, et al. From famine to feast: the role of methylglyoxal production in Escherichia coli. Mol Microbiol. 1998;27:553–562.
- Freedberg WB, Kistler WS, Lin EC. Lethal synthesis of methylglyoxal by Escherichia coli during unregulated glycerol metabolism. J Bacteriol. 1971;108:137–144.
- Rekarte UD, Zwaig N, Istúriz T. Accumulation of methylglyoxal in a mutant of Escherichia coli constitutive for gluconate catabolism. J Bacteriol. 1973;115:727–731.
- Ackerman RS, Cozzarelli NR, Epstein W. Accumulation of toxic concentrations of methylglyoxal by wild-type Escherichia coli K-12. J Bacteriol. 1974;119:357–362.
- Kadner RJ, Murphy GP, Stephens CM. Two mechanisms for growth inhibition by elevated transport of sugar phosphates in Escherichia coli. J Gen Microbiol. 1992;138:2007–2014.
- MacLean MJ, Ness LS, Ferguson GP, et al. The role of glyoxalase I in the detoxification of methylglyoxal and in the activation of the KefB K + efflux system in Escherichia coli. Mol Microbiol. 1998;27:563–571.
- Ferguson GP. Protective mechanisms against toxic electrophiles in Escherichia coli. Trends Microbiol. 1999;7:242–247.
- Misra K, Banerjee AB, Ray S, et al. Glyoxalase III from Escherichia coli: a single novel enzyme for the conversion of methylglyoxal into D-lactate without reduced glutathione. Biochem J. 1995;305: 999–1003.
- Subedi KP, Choi D, Kim I, et al. Hsp31 of Escherichia coli K-12 is glyoxalase III. Mol Microbiol. 2011;81:926–936.
- Misra K, Banerjee AB, Ray S, et al. Reduction of methylglyoxal in Escherichia coli K12 by an aldehyde reductase and alcohol dehydrogenase. Mol Cell Biochem. 1996;156:117–124.
- Saikusa T, Rhee H, Watanabe K, et al. Metabolism of 2-oxoaldehydes in bacteria: purification and characterization of methylglyoxal reductase from Escherichia coli. Agric Biol Chem. 1987;51:1893–1899.
- Ko J, Kim I, Yoo S, et al. Conversion of methylglyoxal to acetol by Escherichia coli aldo-keto reductases. J Bacteriol. 2005;187:5782–5789.
- Abdallah J, Mihoub M, Gautier V, et al. The DJ-1 superfamily members YhbO and YajL from Escherichia coli repair proteins from glycation by methylglyoxal and glyoxal. Biochem Biophys Res Commun. 2016;470:282–286.
- Lee C, Lee J, Lee JY, et al. Characterization of the Escherichia coli YajL, YhbO and ElbB glyoxalases. FEMS Microbiol Lett. 2016;363:fnv239.
- Delpierre G, Collard F, Fortpied J, et al. Fructosamine 3-kinase is involved in an intracellular deglycation pathway in human erythrocytes. Biochem J. 2002;365:801–808.
- Delpierre G, Rider MH, Collard F, et al. Identification, cloning, and heterologous expression of a mammalian fructosamine-3-kinase. Diabetes. 2000;49:1627–1634.
- Gemayel R, Fortpied J, Rzem R, et al. Many fructosamine 3-kinase homologues in bacteria are ribulosamine/erythrulosamine 3-kinases potentially involved in protein deglycation. FEBS J. 2007;274:4360–4374.
- Wiame E, Delpierre G, Collard F, et al. Identification of a pathway for the utilization of the Amadori product fructoselysine in Escherichia coli. J Biol Chem. 2002;277:42523–42529.
- Katz C, Cohen-Or I, Gophna U, et al. The ubiquitous conserved glycopeptidase Gcp prevents accumulation of toxic glycated proteins. MBio. 2010; 1:e00195-10.
- Cohen-Or I, Katz C, Ron EZ. AGEs secreted by bacteria are involved in the inflammatory response. PLoS One. 2011;6:e17974.
- Cohen-Or I, Katz C, Ron EZ. Metabolism of AGEs-bacterial AGEs are degraded by metallo-proteases. PLoS One. 2013;8:e74970.
- Yuan B, Cao H, Jiang Y, et al. Efficient and accurate bypass of N2-(1-carboxyethyl)-2'-deoxyguanosine by DinB DNA polymerase in vitro and in vivo. Proc Natl Acad Sci USA. 2008;105:8679–8684.
- Frischmann M, Bidmon C, Angerer J, et al. Identification of DNA adducts of methylglyoxal. Chem Res Toxicol. 2005;18:1586–1592.
- Schneider M, Thoss G, Hübner-Parajsz C, et al. Determination of glycated nucleobases in human urine by a new monoclonal antibody specific for N2-carboxyethyl-2'-deoxyguanosine. Chem Res Toxicol. 2004;17:1385–1390.
- Li H, Nakamura S, Miyazaki S, et al. N2-carboxyethyl-2'-deoxyguanosine, a DNA glycation marker, in kidneys and aortas of diabetic and uremic patients. Kidney Int. 2006;69:388–392.
- Fuchs RP, Fujii S, Wagner J. Properties and functions of Escherichia coli: Pol IV and Pol V. Adv Protein Chem. 2004;69:229–264.
- McKenzie GJ, Harris RS, Lee PL, et al. The SOS response regulates adaptive mutation. Proc Natl Acad Sci USA. 2000;97:6646–6651.
- Tamae D, Lim P, Wuenschell GE, et al. Mutagenesis and repair induced by the DNA advanced glycation end product N2-1-(carboxyethyl)-2'-deoxyguanosine in human cells. Biochemistry. 2011;50:2321–2329.
- Jarosz DF, Beuning PJ, Cohen SE, et al. Y-family DNA polymerases in Escherichia coli. Trends Microbiol. 2007;15:70–77.
- Boteva E, Handzhiyski Y, Kotseva M, et al. Phosphoglucose isomerase deficiency in Escherichia coli K-12 is associated with increased spontaneous mutation rate. AiM. 2018;08:390–405.
- Gao H, Chen Y, Leary JA. Kinetic measurements of phosphoglucose isomerase and phosphomannose isomerase by direct analysis of phosphorylated aldose–ketose isomers using tandem mass spectrometry. Int J Mass Spectrom. 2005;240:291–299.
- Ishii N, Suga Y, Hagiya A, et al. Dynamic simulation of an in vitro multi-enzyme system. FEBS Lett. 2007;581:413–420.
- Kao HW, Lee SC. Phosphoglucose isomerases of hagfish, zebrafish, gray mullet, toad, and snake, with reference to the evolution of the genes in vertebrates. Mol Biol Evol. 2002;19:367–374.
- Hansen T, Schönheit P. Escherichia coli phosphoglucose isomerase can be substituted by members of the PGI family, the PGI/PMI family, and the cPGI family. FEMS Microbiol Lett. 2005;250:49–53.
- Richarme G, Mihoub M, Dairou J, et al. Parkinsonism-associated protein DJ-1/Park7 is a major protein deglycase that repairs methylglyoxal- and glyoxal-glycated cysteine, arginine, and lysine residues. J Biol Chem. 2015;290:1885–1897.
- Richarme G, Liu C, Mihoub M, et al. Guanine glycation repair by DJ-1/Park7 and its bacterial homologs. Science. 2017;357:208–211.
- Lo TW, Westwood ME, McLellan AC, et al. Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with N alpha-acetylarginine, N alpha-acetylcysteine, and N alpha-acetyllysine, and bovine serum albumin. J Biol Chem. 1994;269:32299–32305.
- Rabbani N, Thornalley PJ. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem Biophys Res Commun. 2015;458:221–226.