
Abstract
This study aimed to investigate the effect of upregulated peroxisome proliferator-activated receptor-gamma (PPAR-γ) activity on cyclooxygenase 2 (COX-2) expression and N-methyl-d-aspartic acid (NMDA)-induced excitatory neurotoxicity in primary cultured cortical neurons. Rat cortical neurons were cultured for 8 days in vitro, and divided into control, NMDA, MK-801 (selective NMDA antagonist), rosiglitazone (ROSI, PPAR-γ agonist), GW9662 (PPAR-γ antagonist), NS398 (selective COX-2 antagonist) and NS398 + ROSI groups. Two hours after treatment in each group, cell viability, intracellular Ca2+ concentrations, PPAR-γ and COX-2 protein expression were detected by CCK-8 assay, flow cytometry and western blot assay, respectively. The results showed that compared with the control group, 100 μmol/L of NMDA significantly decreased the neuronal cell viability, increased Ca2+ concentrations, which also increased the COX-2 protein expression and decreased PPAR-γ expression in neurons. Compared with the NMDA group, the cell viability was increased, Ca2+ concentrations and COX-2 protein expression were significantly decreased, PPAR-γ expression was significantly increased in the MK-801, ROSI, NS398 and ROSI + NS398 groups (both P < 0.01). This finding suggested that upregulation of PPAR-γ activity can inhibit COX-2 expression, decrease Ca2+ concentrations in primary cultured cortical neurons, and protect neurons against NMDA-induced excitatory neurotoxicity.
Introduction
The pathogenesis and prevention of neurodegenerative diseases has become a research hotspot in the field of neuroscience. In the central nervous system, glutamate receptors are classified into ionotropic and metabotropic receptors according to their structural and functional features. The ionotropic glutamate receptors are located in the dense region of the postsynaptic membrane, including N-methyl-d-aspartate (NMDA) receptors and non-NMDA receptors (α-amino-3-hydroxy-5-methylisoxazol-4-propionic acid receptor and kainate receptor). NMDA receptors are ligand- and voltage-gated ion channels, and are involved in the generation and maintenance of synapses between central neurons. Ligand-gated cation channels have high permeability to Ca2+. Excessive activation of NMDA receptors can cause excessive influx of Na+ and Cl−, induce acute osmotic swelling of cells and intracellular Ca2+ overload, activate Ca2+-dependent enzymes, such as neuronal nitric oxide synthase, and cause the production of large amounts of nitric oxide and oxygen-free radicals, mitochondrial damage, and finally leading to neuronal apoptosis or necrosis and the process called excitatory neurotoxicity [Citation1, Citation2].
Cyclooxygenase (COX), or prostaglandin-endoperoxide synthase, is a rate-limitinge nzyme for prostanoid formation. COX has at least two isoforms, COX-1 and COX-2. COX-1 is constitutively expressed in all tissues, and plays an important role in the maintenance of the normal physiological function of the body. COX-2 is an inducible enzyme, which is not expressed or is expressed at extremely low levels in normal tissues, but can be induced by cytokines, growth factors and tumour promoters. COX-2 is involved in the maintenance of synaptic function [Citation3]. Pasinetti and Aisen found that increased neuronal COX-2 expression was associated with the development of chronic neurodegenerative diseases such as Alzheimer's disease [Citation4]. After brain injury, the COX-2 expression in cortical neurons was increased [Citation5]. These findings suggested that COX-2 expression in the brain is associated with neuropathological processes. A study found that COX-2 inhibitor can protect cerebellar granule neurons from glutamate-mediated neurotoxicity [Citation6]. So, in the present study, we hypothesized that increased expression of COX-2 protein is associated with excitatory neurotoxicity induced by overactivation of NMDA receptors in cortical neurons.
Peroxisome proliferator-activated receptors (PPARs) were first discovered by Issemann and Green [Citation7] in 1990. PPARs belong to the steroid hormone receptor superfamily, and have three isoforms (α, δ and γ), which can be activated by peroxisome proliferators and fatty acids. PPARs can regulate the transcription and expression of many genes by binding to specific DNA response elements, which are involved in the regulation of lipid and glucose metabolism, cell differentiation and apoptosis, inflammation and immune responses [Citation8]. PPAR-γ exhibits complex and diverse biological functions. The protective effect of PPAR-γ against cerebral ischemic injury is mainly associated with its inhibitory effect on the secretion of tumour necrosis factor-α (TNF-α), COX-2 and inducible nitric oxide synthase (iNOS) [Citation9]. The aim of the study was to investigate the role of upregulation of PPAR-γ activity on COX-2 protein expression and NMDA-induced excitatory neurotoxicity in rat cortical neurons.
Materials and methods
Experimental animals
Sixty neonatal Wistar rats aged 1 day were purchased from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China, license number: SCXK[Hu]2012-0002).
Ethics statement
All experimental procedures were approved by the Animal Ethics Committee of Hainan Medical University.
Drugs and reagents
Neurobasal-A medium, fetal bovine serum (Grand Island Biological Co. GIBCO, USA); poly-lysine, NS398, rosiglitazone (ROSI), NMDA, GW9662, MK-801 (Sigma, USA); microtubule-associated protein 2 antibody (MAP2, Abcam, USA); B27 (Life Technologies Ltd., USA); Cell Counting Kit-8 (CCK-8) (Dojindo, Japan); polyvinylidene fluoride membrane (Millipore Corp., USA); COX-2 (H-62) polyclonal antibody, PPAR-γ (H-100) polyclonal antibody (Santa Cruz Biotechnology, USA); glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody, horseradish peroxidase-conjugated goat anti-rabbit IgG (Bioworld Technology Inc., USA).
Primary culture of rat cortical neurons
Neonatal rats were decapitated under aseptic conditions. Cerebral cortex was obtained, meninges, blood vessels and hippocampus were then removed. Cerebral cortex was cut into 1 mm3 blocks in 4 °C D-Hank’s solution (pH 7.0), and digested in an equal volume of 0.25% typsin (with ethylenediaminetetraacetic acid) in a 37 °C water bath for 30 minutes with gentle shaking every 2 minutes. DMEM containing 10% fetal bovine serum was added to terminate trypsin digestion. After the samples were gently blown and beaten, the tube was allowed to stand at low temperature. The supernatant was collected and filtered through a 100–200-μm nylon mesh into 50-mL centrifuge tubes. Then the cell suspension was centrifuged at 1000 g for 6 minutes. After the supernatant was removed, precipitated cells were resuspended in Neurobasal-A medium containing 10% fetal bovine serum. Cells at the density of 5 × 105 cells/mL were seeded on the poly-l-lysine-coated flasks or plates, and incubated in a 5% CO2 incubator at 37 °C for 24 hours. The medium was replaced by serum-free Neurobasal-A medium containing 2% B27. Cytarabine (10 μmol/L) was added to suppress the proliferation of glial cells and non-neuronal cells. One-half of the medium was replaced every 2 days. Primary cortical neurons were cultured for 8 days in vitro.
Immunofluorescence detection of the primary cultured rat cortical neurons
After removal of the medium, primary cultured neurons were washed with phosphate buffered saline (PBS) three times for 5 minutes each, and fixed in 4% paraformaldehyde for 30 minutes at room temperature, followed by three washes in PBS for 5 minutes each. Then the neurons were permeabilized with 0.5% Triton X-100 solution at room temperature for 10 minutes, washed in PBS three times for 5 minutes each. After being blocked with 5% bovine serum albumin at 37 °C for 2 hours, the neurons were incubated with primary antibody (1:200) at 4 °C overnight. The wet box was then taken out and equilibrated at room temperature for 30 minutes, followed by three washes in PBS for 5 minutes each. Then the neurons were incubated with FITC-conjugated Dnk anti-rabbit secondary antibody (1:400) at 37 °C for 1 hour in the dark, and washed with PBS three times with 5 minutes each. Cell nuclei were stained with DAPI at room temperature for 15 minutes, and washed in PBS three times for 5 minutes each. After mounting, the cells were then observed and photographed using fluorescence microscopy.
Grouping
The cultured cortical neurons were divided into control group (normal control group), NMDA group (neurons were treated with 100 μmol/L NMDA and 10 μmol/L glycine for 2 hours), MK-801 group (neurons were treated with100 μmol/L NMDA, 10 μmol/L glycine and 10 μmol/L MK-801 for 2 hours), ROSI group (neurons were treated with 1 μmol/L ROSI for 30 minutes, and then treated with 100 μmol/L NMDA for 2 hours), GW9662 group (neurons were treated with 1 μmol/L GW9662 for 30 minutes, and then treated with NMDA 100 μmol/L for another 2 hours), NS398 group (neurons were treated with 30 μmol/L NS398 for 30 minutes, and then treated with 100 μmol/L NMDA for another 2 hours), NS398 + ROSI group (neurons were treated with ROSI and 30 μmol/L NS398 for 30 minutes, and then treated with 100 μmol/L NMDA for another 2 hours). MK-801 is a selective NMDA antagonist. ROSI is a PPAR-γ agonist. GW9662 is a PPAR-γ antagonist. NS398 is a selective COX-2 antagonist.
Determination of cell viability using CCK-8 assay
Two hours after treatment in each group, cultures were washed with pre-warmed Mg2+-free Locke’s solution; 110 μL medium containing 10 μL CCK-8 were added into each well, the cultures were then incubated at 37 °C in the dark for 2 hours. A blank control well was set. Optical density (OD) values were measured at 450 nm wavelength in each well.
Western blot assay
Neuronal cells were harvested, and lysed in 1 mL lysis buffer containing 10 μL phenylmethylsulfonyl fluoride (PMSF) solution at 4 °C for 30 minutes. The samples were centrifuged at 12,000 rpm at 4 °C for 10 minutes. Protein concentration was quantified using the bicinchoninic acid assay (BCA). OD values were measured at 562 nm wavelength. Protein concentrations of samples were calculated based on the standard curve. Polyacrylamide gel electrophoresis was conducted using 10% separating gel and 5% stacking gel. Proteins were transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were then blocked and incubated with primary antibody PPAR-γ (H-100) and COX-2 (H-62), followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit IgG. The protein membranes were visualized with an enhanced chemiluminescence reagent.
Detection of Ca2+ concentrations using flow cytometry
Cells from each group were taken, washed with HBSS, and centrifuged at 251.55 g for 5 minutes. Cells were resuspended with 2.5 μmol/L Fluo 4-AM working solution to a final concentration of 100 μL, incubated at 37 °C in the dark for 30 minutes. After washing three times with HBSS, cells were centrifuged at 1500 rpm for 5 minutes. The mean fluorescence intensity was estimated at the FL-1 channel.
Statistical analysis
Data were collected in at least five independent experiments. Results were analyzed with SPSS 13.0 statistical software, and expressed as mean values with standard deviation (± SD). Differences between groups were compared using one-way analysis of variance followed by Dunnett’s test. A P-value of less than 0.05 was considered to indicate a statistically significant difference.
Results and discussion
Identification of primary cultured rat cortical neurons
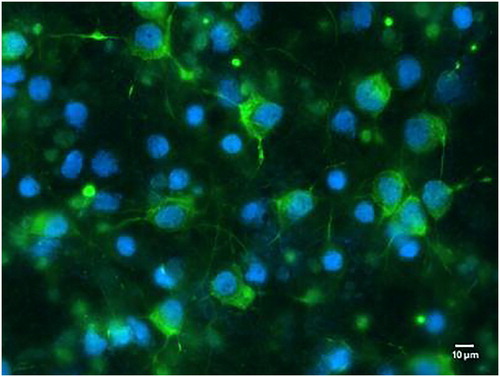
Fluorescence microscopy showed that neurons expressed MAP2, which presented green fluorescence. The nuclei displayed blue fluorescence. Neuronal cells showed scattered or massive growth. Neuronal processes exhibited obvious reticulated distribution ().
Figure 1. Primary cultured neurons in vitro (× 400). Green fluorescence revealed microtubule-associated protein 2 antibody (MAP2) expression; the nuclei were stained blue.
Upregulation of PPAR-γ activity inhibits NMDA-induced reduction of neuronal cell viability in cultured cortical neurons
The overactivation of the NMDA receptor can trigger a Ca2+ influx, which contributes to neuronal apoptosis or necrosis [Citation10]. NMDA-induced excitatory neurotoxicity is considered to be strongly associated with the development of neurodegenerative diseases. Therefore, inhibition of excitatory neurotoxicity induced by overactive NMDA receptors is important for the treatment of neuronal damage and various neurodegenerative diseases.
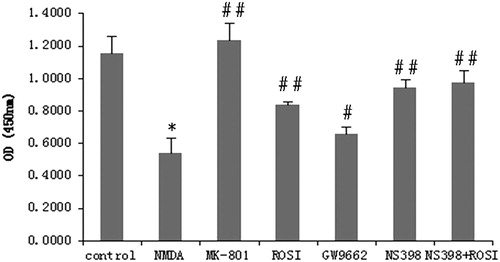
In the present study, we used CCK-8 assay to detect cell viability of each group. The results showed that compared with the control group, the neuronal cell viability was significantly reduced in the NMDA group (P < 0.01), indicating that 100 μmol/L NMDA significantly decreased the neuronal cell viability. MK-801 is a selective NMDA antagonist, which can decrease NMDA-induced neurotoxicity. There was no significant difference between the MK-801 and control groups. Compared with the NMDA group, cell viability was significantly increased in the ROSI, NS398 and ROSI + NS398 groups (P < 0.01).
ROSI is an insulin sensitiser, which belongs to the class of thiazolidinediones (TZDs), and acts as a more effective agonist of PPAR-γ [Citation11], and GW9662 is a PPAR-γ antagonist. The results showed that cell viability was decreased in the GW9662 group compared with the ROSI group, which was increased in the GW9662 group than that in the NMDA group (P < 0.05) (). The results indicated that ROSI treatment can inhibit NMDA-induced neurotoxicity and reduce neuronal damage through upregulation of PPAR-γ activity.
Figure 2. Changes in neuronal cell viability in each group. Compared with control group, NMDA significantly reduced neuronal cell viability. Compared with NMDA group, MK-801(selective NMDA antagonist), rosiglitazone (ROSI, PPAR-γ agonist), and NS398 (selective COX-2 antagonist) both significantly increased cell viability. GW9662 (PPAR-γ antagonist) decreased the cell viability. *P < 0.01, vs. control group; #P < 0.05, ##P < 0.01, vs. NMDA group.
Upregulation of PPAR-γ activity decreases Ca2+ concentration in neurons with NMDA-induced neurotoxicity
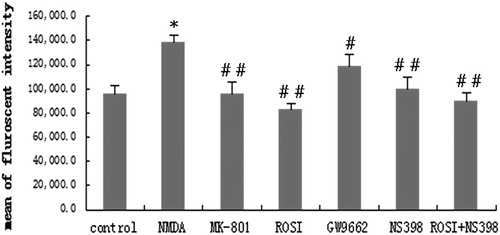
Intracellular Ca2+ concentration of each group was detected by flow cytometry analysis. As shown in , compared with the control group, the intracellular Ca2+ concentration was significantly increased in the NMDA group (P < 0.01). MK-801 suppressed NMDA-induced neurotoxicity, and Ca2+ concentrations were significantly reduced in the MK-801 group compared to the NMDA group (P < 0.01). Intracellular Ca2+ concentrations were significantly decreased in the ROSI, NS398 and ROSI + NS398 groups compared with the NMDA group (P < 0.01). GW9662 reduced PPAR-γ activity. There were significant differences in the Ca2+ concentrations between the GW9662 and NMDA groups (P < 0.05). These results indicated that intracellular Ca2+ concentrations were increased in neurons with NMDA-induced neurotoxicity, and upregulation of PPAR-γ activity can decrease intracellular Ca2+ concentrations in neurons.
Figure 3. Changes in intracellular Ca2+ in each group. Compared with control group, NMDA significantly increased intracellular Ca2+ concentration. Compared with NMDA group, MK-801(selective NMDA antagonist), rosiglitazone (ROSI, PPAR-γ agonist), and NS398 (selective COX-2 antagonist) both significantly decreased intracellular Ca2+ concentration. GW9662 (PPAR-γ antagonist) increased the cell viability. *P < 0.01, vs. control group; #P < 0.05, ##P < 0.01, vs. NMDA group.
Upregulation of PPAR-γ inhibits NMDA-induced neurotoxicity through downregulating COX-2 protein expression
COX-2 can catalyze the conversion of arachidonic acid into thromboxane A2, prostaglandin E2 and prostaglandin H2, which is associated with neuroinflammation, and contributes to the progression of neurodegenerative diseases [Citation12]. A recent study had shown that COX-2 expression depends on the activation of calcineurin, which is associated with increased Ca2+ concentration in the cytoplasm of neurons [Citation13]. Selective inhibition of the binding of neuronal nitric oxide synthase to COX-2 can suppress NMDA-induced excitatory neurotoxicity and exert neuroprotective properties [Citation14, Citation15].
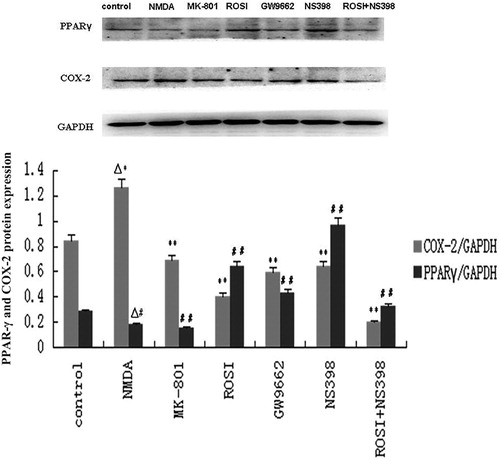
Western blot assay was used to determine the protein expression of PPARγ and COX-2 in neurons of each group. Results showed that compared with the control group, COX-2 protein expression was significantly increased, while PPARγ protein expression was decreased in the NMDA group (P < 0.01). Compared with the NMDA group, COX-2 protein expression was significantly decreased in MK-801 group (P < 0.01); PPARγ protein expression was increased and COX-2 protein expression was decreased in the ROSI, NS398, and ROSI + NS398 groups (P < 0.01). PPARγ protein expression was decreased, and COX-2 protein expression was increased in the GW9662 group compared with the ROSI group (P < 0.01). These results indicated that NMDA-induced neurotoxicity was associated with the increase in COX-2 protein expression, and increased PPARγ activity could downregulate COX-2 protein expression in neurons with NMDA-induced neurotoxicity ().
Figure 4. Western blot assay of PPAR-γ and COX-2 protein expression in neurons of each group. Compared with control group, NMDA significantly increased COX-2 protein expression, and decreased PPAR-γ expression. Compared with NMDA group, MK-801(selective NMDA antagonist), rosiglitazone (ROSI, PPAR-γ agonist), and NS398 (selective COX-2 antagonist) both significantly decreased COX-2 protein expression, and increased PPAR-γ expression. GW9662 (PPAR-γ antagonist) increased COX-2 protein expression, and decreased PPAR-γ expression. ▵P < 0.01, vs. control group; **P < 0.01, ##P < 0.01, vs. NMDA group.
ROSI exerted neuroprotective effect through reducing the COX-2 expression surrounding the infarcted site of the ischemic hemisphere [Citation16–18]. Du et al. [Citation19] found that PPAR-γ-mediated inhibition of COX-2 expression can protect hippocampal neurons from damage. The protective effect of hyperbaric oxygen preconditioning against hypoxia/hypoglycemia-induced injury of primary cultured cortical neurons is also achieved through activating PPAR-γ expression and increasing antioxidant enzyme activity [Citation20]. Selective COX-2 inhibitor NS-398 and endogenous PPAR-γ ligand can act synergistically and exert neuroprotective effects on rat pheochromocytoma cells [Citation21]. In the present study, we found that combination of ROSI and selective COX-2 inhibitor NS-398 not only decreased Ca2+ concentrations, increased neuronal cell viability, but also increased PPAR-γ protein expression and decreased COX-2 protein expression in cultured neurons, indicating that decreased PPAR-γ activity and increased COX-2 protein expression are associated with excitotoxic neuronal damage induced by NMDA in cultured cortical neurons. The combination of ROSI and NS-398 can inhibit NMDA-induced excitatory neurotoxicity in primary cultured cortical neurons.
Conclusions
Our results suggested that upregulation of PPARγ activity can increase the neuronal cell viability, and protect cortical neurons against NMDA-induced excitatory neurotoxicity through inhibiting COX-2 expression and reducing Ca2+ concentrations in cultured cortical neurons.
Disclosure Statement
The authors report no conflicts of interest.
Additional information
Funding
Related Research Data
References
- Prentice H, Modi JP, Wu JY. Mechanisms of neuronal protection against excitotoxicity, endoplasmic reticulum stress, and mitochondrial dysfunction in stroke and neurodegenerative diseases. Oxid Med Cell Longev. 2015;2015:1.
- Lewerenz J, Maher P. Chronic glutamate toxicity in neurodegenerative diseases-what is the evidence?. Front Neurosci. 2015;9:469.
- Vaughan CW. Enhancement of opioid inhibition of GABAergic synaptic transmission by cyclo-oxygenase inhibitors in rat periaqueductal grey neurones. Br J Pharmacol. 1998;123:1479–1481.
- Pasinetti GM, Aisen PS. Cyclooxygenase-2 expression is increased in frontal cortex of Alzheimer's disease brain. Neuroscience. 1998;87:319–324.
- Hirst WD, Young KA, Newton R, et al. Expression of COX-2 by normal and reactive astrocytes in the adult rat central nervous system. Mol Cell Neurosci. 1999;13:57–68.
- Strauss KI, Marini AM. Cyclooxygenase-2 inhibition protects cultured cerebellar granule neurons from glutamate-mediated cell death. J Neurotrauma. 2002;19: 627–638.
- Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650.
- D'Angelo M, Antonosante A, Castelli V, et al. PPARs and Energy Metabolism Adaptation during Neurogenesis and Neuronal Maturation. Int J Mol Sci. 2018;19:E1869.
- Su M, Cao J, Huang J, et al. The in vitro and in vivo anti-inflammatory effects of a phthalimide PPAR-γ agonist. Mar Drugs. 2017;15:7.
- Paoletti P. Molecular basis of NMDA receptor functional diversity. Eur J Neurosci. 2011;33:1351–1365.
- Bo QL, Chen YH, Yu Z, et al. Rosiglitazone pretreatment protects against lipopolysaccharide-induced fetal demise through inhibiting placental inflammation. Mol Cell Endocrinol. 2016;423:51–59.
- Sil S, Ghosh T. Role of cox-2 mediated neuroinflammation on the neurodegeneration and cognitive impairments in colchicine induced rat model of Alzheimer's Disease. J Neuroimmunol. 2016;291:115–124.
- Chetsawang J, Nudmamud-Thanoi S, Phonchai R, et al. Methamphetamine toxicity-induced calcineurin activation, nuclear translocation of nuclear factor of activated T-cells and elevation of cyclooxygenase 2 levels are averted by calpastatin overexpression in neuroblastoma SH-SY5Y cells. Neurotoxicology. 2018;67:287–295.
- Tian J, Kim SF, Hester L, et al. S-nitrosylation/activation of COX-2 mediates NMDA neurotoxicity. Proc Natl Acad Sci USA. 2008;105:10537–10540.
- Stark DT, Bazan NG. Synaptic and extrasynaptic NMDA receptors differentially modulate neuronal cyclooxygenase-2 function, lipid peroxidation, and neuroprotection. J Neurosci. 2011;31:13710–13721.
- Mu Q, Wang L, Hang H, et al. Rosiglitazone pretreatment influences thrombin-induced phagocytosis by rat microglia via activating PPARγ and CD36. Neurosci Lett. 2017;651:159–164.
- Villapol S. Roles of peroxisome proliferator-activated receptor gamma on brain and peripheral inflammation. Cell Mol Neurobiol. 2018;38:121–132.
- Cai W, Yang T, Liu H, et al. Peroxisome proliferator-activated receptor γ (PPARγ): A master gatekeeper in CNS injury and repair. Prog Neurobiol. 2018;163-164:27–58.
- Du H, Chen X, Zhang J, et al. Inhibition of COX-2 expression by endocannabinoid 2-arachidonoylglycerol is mediated via PPAR-γ. Br J Pharmacol. 2011;163:1533–1549.
- Zeng Y, Xie K, Dong H, et al. Hyperbaric oxygen preconditioning protects cortical neurons against oxygen-glucose deprivation injury: role of peroxisome proliferator-activated receptor-gamma. Brain Res. 2012;1452:140–150.
- Abraki SB, Khalaj L, Shaerzadeh F, et al. Simultaneous inhibition of COX-2 and activation of PPAR-γ resulted in the same level and pattern of neuroprotection as they were targeted separately. J Mol Neurosci. 2013;49: 116–129.