
Abstract
Chlamydia trachomatis is an important human pathogen responsible for a variety of important diseases. pORF5, a plasmid protein in C. trachomatis, has been reported to contribute to chlamydial pathogenesis. Apoptosis and autophagy are two important cellular processes, which also participate in the pathological and the physiological processes in C. trachomatis infection. The present study aimed to explore the effects of pORF5 protein on autophagy and apoptosis in HeLa cells. Stable pORF5-HeLa and vector-HeLa cell lines that were transduced with pORF5 recombination lentivirus and control lentivirus, respectively, were constructed. Immunofluorescence was used to observe the cells. Western blotting was carried out to detect expression of target proteins. Real-time polymerase chain reaction was used to determine the mRNA expression. Flow cytometry, TUNEL assay and Hoechst staining were employed to detect apoptosis. pORF5 sensitized HeLa cells to autophagy that was induced by nutrient deprivation. In addition, pORF5 increased the ratio of LC3-II/LC3-I and Beclin-1 expression, down-regulated the expression of Bax and Caspase-3 and up-regulated Bcl-2 expression to suppress apoptosis induced by TNF-α. Autophagy inhibitor 3-MA elevated apoptotic rate significantly in pORF5-HeLa cells. The study preliminarily confirms that plasmid protein pORF5 mediates the crosstalk between autophagy and apoptosis, which may contribute to persistent C. trachomatis infection in host cells.
Introduction
Chlamydia trachomatis is a primary human pathogen associated with several important human diseases such as trachoma, arthritis and sexually transmitted diseases [Citation1–3], and it is also reported to significantly increase the risk of HIV infection and cervical carcinoma [Citation4, Citation5]. Since urogenital chlamydial infections are frequently asymptomatic, most infected people do not receive timely medical treatment, and a substantial proportion of untreated cases develop long-term complications, including pelvic inflammatory disease, which has become a serious threat to human health. New approaches to the treatment and prevention of chlamydial infection need to be developed. However, the molecular mechanisms of chlamydial pathogenesis are not fully elucidated.
Almost all clinical isolates of C. trachomatis maintain a highly conserved plasmid of approximately 7.5 kb, which encodes eight open reading frames (ORFs). Chlamydia trachomatis plasmid is a virulence factor in the pathogenesis of genital tract infection in female pig-tailed macaques [Citation6]. Plasmid-deficient C. trachomatis significantly reduces pathogenicity in ocular tissues of primates and genital tract tissues of mice [Citation7–10]. pORF5 (Pgp3), encoded by one of the eight ORFs, is the only plasmid-secreted protein mainly distributed in the cytosol of host cells [Citation11]. Previous studies have shown that pORF5 could effectively induce the release of inflammatory cytokines and promote chlamydial ascending infection by neutralizing anti-chlamydial activity of antimicrobial peptide LL-37 [Citation12–15], indicating that pORF5 plasmid protein plays an important role in the pathogenesis of C. trachomatis.
Chlamydia trachomatis is an obligate intracellular pathogen that relies on nutrients from the host cells for growth and multiplication. Autophagy is a lysosomal recycling process, which degrades the captured proteins, lipids and carbohydrates to provide nutrients. Several intracellular pathogens, including Salmonella and respiratory syncytial virus, have been demonstrated to evolve strategies to extract the products of autophagy [Citation16–20]. A previous study showed that C. trachomatis induces autophagy, and autophagy inhibitor 3-MA has significant negative effects on infection and development of C. trachomatis [Citation21], suggesting a potential role of host autophagy in intracellular growth and persistent infection by C. trachomatis.
Apoptosis, a primary death mechanism of cells, is closely linked to autophagy [Citation22–25]. Cumulative evidence indicates that autophagy serves as an antagonist to block apoptotic cell death by promoting cell survival. In turn, apoptosis can also inhibit autophagy. For example, apoptotic signaling leads to activation of caspases, and activated caspases can cleave autophagy-regulator proteins, such as Beclin-1, to inactivate autophagy and promote apoptosis [Citation26–28]. In addition, several other pro-apoptotic proteins including Bak, BNIP3 and Nix, have been identified as regulators of autophagy by disrupting the interaction between Bcl-2 and Beclin-1 to suppress autophagy [Citation29, Citation30]. Some evidence shows that C. trachomatis has evolved effective strategies to inhibit apoptosis during the course of infection [Citation31–34]. Although C. trachomatis can mediate autophagy and apoptosis resistance, the mechanisms by which the two important cellular processes control cell survival and cell death are not completely understood yet.
In the present study, we first investigate the effects of autophagy and apoptosis on HeLa cells triggered by pORF5 plasmid protein of C. trachomatis and then detect the expression of autophagy-related proteins (microtubule-associated protein 1 light chain 3 (LC-3) and Beclin-1) and apoptotic proteins (Bcl-2, Bax and Caspase-3) during autophagy and apoptosis to examine the molecular mechanism, by which pORF5 regulates autophagy and apoptosis.
Materials and methods
Reagents
The antibodies to LC3A/B, Beclin-1, Bax and Bcl-2 were acquired from Cell Signaling Technology (Beverly, MA). The antibody to GAPDH, Cy3-conjugated IgG secondary antibody and peroxidase- conjugated secondary antibody were purchased from Proteintech (Chicago, IL). 3-MA, Hoechst 33342 and Bafilomycin A1 (BafA) were obtained from Sigma-Aldrich (St. Louis, MO).
Cells
Stable pORF5-HeLa and vector-HeLa cell lines that were transduced with pORF5 recombination lentivirus and control lentivirus, respectively, were constructed as described previously [Citation35] and kept in our lab. pORF5-HeLa cells and vector-HeLa cells were sustained in DMEM medium (HyClone, Logan, UT) containing 10% (vol/vol) fetal bovine serum (Gibco, Carlsbad, CA) at 37 °C under humidification and 5% CO2 conditions. For stimulation experiments, the two cell lines were grown in 6-well flat-bottom plates (Corning, New York, NY) at a density of 1 × 106/mL for Western blotting and real-time PCR analyses or 24-well flat-bottom plates (Corning) for immunofluorescence analysis. After reaching 70–80% confluence, cells were pre-treated with TNF-α (20 ng/mL), 3-MA (2 mmol/L), BafA (100 nmol/L) or serum starvation as indicated in individual experiments.
Indirect immunofluorescence
The two stable cell lines were cultured on coverslips in 24-well plates for 24 h. After treatment with TNF-α for 6 h, 3-MA or serum starvation for 24 h, the cells were washed with phosphate-buffered saline (PBS) for three times and subsequently fixed for 30 min with 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO) at room temperature. After washing with PBS, permeabilization was performed with 0.1% Triton-X100 (Sigma-Aldrich) in PBS for 10 min. After blocking with 2.5% bovine serum albumin (BSA) at 37 °C for 2 h, the coverslips were incubated with various primary rabbit antibodies at 4 °C overnight. Then, signals were visualized with Cy3-conjugated IgG secondary antibody for 2 h, and nucleuses were stained with DAPI. Digital images were taken with an inverted fluorescence microscope (Nikon, TE2000-S, Japan), and fluorescence intensity was analyzed using Image J software (Media Cybernetics, Silver Spring, MD).
Western blotting
Cells were harvested after treatment and resuspended in RIPA (Beyotime Biotechnology, Shanghai, China) lysis buffer supplemented with protease inhibitor phenylmethanesulfonyl fluoride (Beyotime Biotechnology) for 30 min. Suspensions of lysed cells were then centrifuged at 1000 rpm at 4 °C for 10 min. Protein concentrations were determined with Bio-Rad protein assay system (Bio-Rad, Hercules, CA). Aliquots of cell extracts containing 30 μg of protein were subjected to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred to a polyvinylidene fluoride membrane (Millipore, Billerica, MA). After blocking with 5% non-fat milk for 1 h at room temperature, the membranes were immunoblotted with various primary antibodies at 4 °C overnight. Then, goat anti-rabbit peroxidase-conjugated secondary antibody was used for incubation at 37 °C for 1 h. The protein signals were detected by ECL Western blot detection reagents (Thermo Fisher Scientific, Waltham, MA). β-Actin was used as the loading control.
Flow cytometry
Cells were trypsinized and washed with ice-cold PBS for three times. Aliquots of cells (5 × 105) were resuspended in 500 μL of binding buffer and stained with Annexin-V-APC and 7-aminoactinomycin D (7-AAD) (BD Biosciences, San Diego, CA) according to the manufacturer's instructions. The percentages of apoptotic cells were determined by a flow cytometer (Beckman Coulter, Fullerton, CA) with CellQuest software (BD Biosciences).
Real-time PCR
Genes that encode apoptosis and autophagy-related proteins were selected as candidate genes for the assessment of their mRNA expression levels under different conditions (). Total RNA was isolated from cells using Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Then, the RNA was used as templates for reverse transcription using TaqMan reverse transcription kit (Applied Biosystems, Foster City, CA). Real-time PCR was performed using TaqMan universal PCR master mix (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. The results were expressed as relative levels of mRNA after being normalized with endogenous control glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Table 1. Primer sequences used in real-time PCR.
Terminal deoxynucleotidyl transferase-mediated dUTP-fluorescein nick end labeling (TUNEL) assay and Hoechst staining
Apoptotic cells were detected by TUNEL assay (Promega, Madison, WI) and Hoechst staining according to manufacturer's instructions. Briefly, cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton-X-10 and then labeled with fluorescein-dUTP for 1 h or Hoechst 33258 for 20 min. Quantification was performed by counting apoptotic cells from five randomly selected fields. Apoptotic rate was evaluated as the ratio of number of apoptotic cells to total cell count × 100.
Statistical analysis
All data were expressed as means ± deviation (SD). Groups were compared by one-way analysis of variance (ANOVA) with Student–Newman–Keuls test. SPSS 19.0 statistical software package (SPSS, Chicago, IL) was used for all statistical analyses. p value < .05 was regarded as statistically significant.
Results and discussion
pORF5 sensitizes HeLa cells to autophagy that is induced by nutrient deprivation
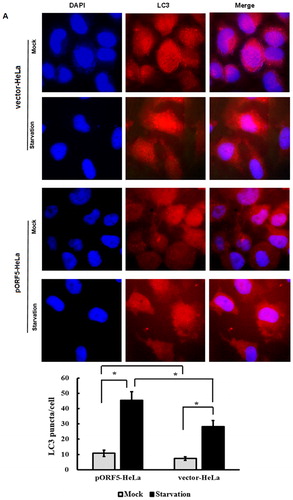
To determine the role of pORF5 plasmid protein of C. trachomatis in autophagy, we first compared punctate LC3, a specific biochemical marker for autophagy, in the two stable cell lines under nutrient starvation condition. Indirect immunofluorescence assay showed that the number of LC3 fluorescence puncta in pORF5-HeLa cells was increased significantly when compared with vector-HeLa cells under serum starvation condition for 24 h (p < .01) (). Quantification showed that there were approximately 45.5 and 28.3 puncta/cell in pORF5-HeLa and vector-HeLa cells, respectively. No significant difference was observed between them under non-serum starvation (10.8 ± 2.1 vs 7.3 ± 1.2) (p > .05).
Figure 1. LC3 puncta analysis in pORF5-HeLa cells and vector-HeLa cells. (A) Signals of LC3 protein in pORF5-HeLa cells and vector-HeLa cells treated either with or without serum starvation for 24 h under a fluorescence microscope (1000×). (B) Quantification of LC3 puncta from a total of 500 cells per treatment from three independent experiments. Note: (A) Red signals represented LC3, and nucleus was stained with DAPI (blue); (B) *p < .01 and #p > .05.
Autophagosome formation is regulated by more than 30 autophagy-related proteins (Atg). Beclin-1 is one of the upstream molecules for recruiting other autophagy proteins to initiate the autophagic pathway. LC3, a key regulator involved in forming autophagosomes, exists in two forms, the cytosolic form (LC3-I) and the lipidated form LC3-II, which resides in the autophagosomal membranes. LC3-II has been widely used as a marker protein for assessing autophagosome formation [Citation36, Citation37]. To determine LC3 and Beclin-1 expression in vector-HeLa and pORF5-HeLa cells after serum starvation for 24 h, LC3 and Beclin-1 mRNA were detected by real-time PCR. The data showed a remarkable increase of LC3 and Beclin-1 mRNA expression in all serum starvation groups. The mRNA levels of LC3 and Beclin-1 in pORF5-HeLa cells treated with serum-free media were 3.1 times (p < .01) and 0.9 times (p < .05), respectively, higher than that in the same cells incubated with serum medium, and 2.0 times (p < .01) and 1.6 times (p < .01), respectively, higher than that in vector-HeLa cells (). Western blotting showed that the ratios of LC3-II/LC3-I in serum starvation groups were significantly higher than those in corresponding groups incubated with serum media (p < .01), especially in pORF5-HeLa cells, indicating that conversion of LC3-I to LC3-II was induced by nutrient deprivation (). Moreover, Beclin-1 expression was also markedly increased after starvation treatment (p < .05) (). The levels of LC3-II/LC3-I and Beclin-1 were increased by 52.2% and 57.8%, respectively, in vector-HeLa, and by 76.6% and 87.2%, respectively, in pORF5-HeLa when compared with the same cells incubated with serum media (p < .05). Meanwhile, the ratio of LC3-II/LC3-I and Beclin-1 expression were increased by 53.3% (p < .01) and 28.7% (p < .05), respectively, in pORF5-HeLa, when compared with vector-HeLa, whereas no significant difference was found in two cell lines under non-starvation condition (p > .05) ().
Figure 2. Effect of pORF5 protein on autophagy under conditions of nutrient deprivation. (A) Protein expression of LC3-I, LC3-II and Beclin-1 in pORF5-HeLa cells and vector-HeLa cells treated under different conditions (serum starvation and non-serum starvation) detected by Western blotting. (B) Effect of pORF5 on protein expression of LC3 and Beclin-1. (C) Bar graph representing the LC3-II/LC3-I ratio. (D) Bar graph representing Beclin-1 expression. (E) Effect of BafA on the ratio of LC3-II/LC3-I. (F) LC3-II accumulation elevation induced by pORF5 in the presence of BafA. Note: Relative protein abundance of each band was normalized to the grey value of β-actin, and the bands were quantified by densitometry expressed as means ± standard deviation. These data represent three independent experiments. *p < .01, **p < .05 and #p > .05.
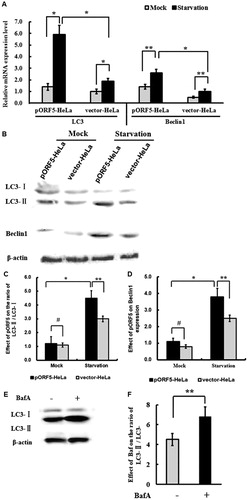
To further validate the effect of pORF5 on autophagy flux, a pharmacological inhibitor of autophagy, which blocks the fusion of autophagosomes with lysosomes, BafA was used to block the flow of autophagy. The data showed that pORF5 elevated LC3-II accumulation in the presence of BafA (p < .05) (), indicating that the conversion of LC3-I to LC3-II was an active process. These results suggest that pORF5 sensitizes HeLa cells to autophagy that is induced by nutrient deprivation.
pORF5 negatively regulates apoptosis of HeLa cells that is induced by TNF-α
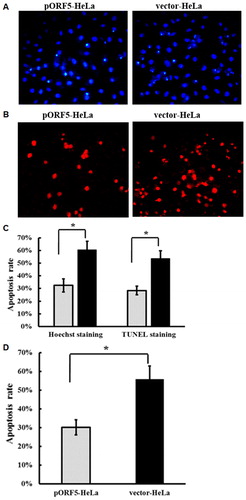
Apoptosis is an active process of cellular suicide trigged by a variety of stress and physiological stimuli. Chlamydia trachomatis has developed different strategies to inhibit host cell apoptosis, and the induction of apoptosis resistance is thought to be an important immune escape mechanism allowing C. trachomatis to replicate inside the host cells [Citation38, Citation39]. We next investigated the role of pORF5 on apoptosis. After treatment with TNF-α, HeLa cells underwent Hoechst staining () and TUNEL assay (), and morphological changes of the cells were observed under a fluorescence microscope, where apoptotic cells showed nuclear chromatin condensation, fragmentation and cell shrinkage. Hoechst staining showed that pORF5 significantly decreased the numbers of cells with condensed chromatin induced by TNF-α (), and TUNEL assay showed that the apoptotic rate of pORF5-HeLa cells was significantly lower than that of vector-HeLa cells (p < .01) (). We also used Annexin-V-APC/7-AAD staining and flow cytometry to determine the extent of apoptosis. The data showed that the apoptosis rate of pORF5-HeLa cells was significantly lower than that of vector-HeLa cells (p < .01) (). The results suggest that pORF5 inhibits TNF-α-induced cell apoptosis.
Figure 3. Inhibitory effect of pORF5 protein on HeLa cells apoptosis induced by TNF-α. (A) Hoechst staining of pORF5-HeLa cells and vector-HeLa cells treated with TNF-α for 6 h. (B) TUNEL staining of pORF5-HeLa cells and vector-HeLa cells treated with TNF-α for 6 h under a fluorescence microscope (200×). (C) Quantification of apoptosis rate performed by counting apoptotic cells from five randomly selected fields, and a significant increase in pORF5-HeLa cells when compared to vector-HeLa cells. (D) Percentage of apoptotic cells detected by flow cytometry in pORF5-HeLa and vector-HeLa. Note: All data are expressed as means ± standard deviation calculated from at least three independent experiments. These data represent three independent experiments. *p < .01.
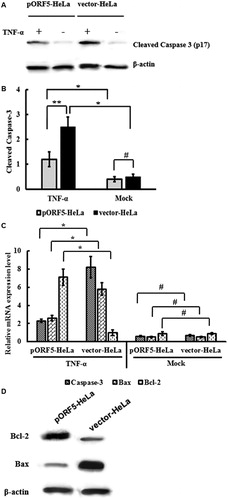
pORF5 plasmid protein inhibits caspase-3 activation and regulates the expression of Bcl-2 and Bax
Apoptosis is tightly regulated by anti-apoptotic and pro-apoptotic effector molecules, which mainly include proteins of the Bcl-2 family and caspases. Bax and Bcl-2 are representative members of the Bcl-2 family, and Caspase-3 is generally considered an important effector protease that is cleaved and activated during apoptosis. To test whether inhibition of Caspase-3 activation was involved in apoptosis resistance that was induced by pORF5, the expression of cleaved p17 subunit of Caspase-3 was detected using Western blotting. The data showed that the expression of p17 subunit of Caspase-3 in pORF5-HeLa cells was significantly lower than that in vector-HeLa cells (p < .05) (). Consistently, the expression of Caspase-3 mRNA in pORF5-HeLa cells was significantly lower than that in vector-HeLa cells (p < .01) (). Moreover, Bax gene expression in pORF5-HeLa cells was significantly reduced, and Bcl-2 gene expression in pORF5-HeLa cells was significantly increased than that in vector-HeLa cells group (p < .01) (). Western blotting showed that Bax protein expression in pORF5-HeLa cells was significantly lower than that in vector-HeLa cells, while Bcl-2 protein expression in pORF5-HeLa cells was significantly higher than that in vector-HeLa cells (p < .01) (). The results indicate that pORF5 plasmid protein inhibits Caspase-3 activation and regulates the expression of Bax and Bcl-2.
Figure 4. Inhibition of caspase-3 activation and regulation of of Bcl-2 and Bax expression caused by pORF5. (A) Western blotting for detection of the expression of cleaved Caspase-3, Bax and Bcl-2 proteins in pORF5-HeLa cells and vector-HeLa cells treated with TNF-α for 6 h. (B) Quantification of p17 Caspase-3 subunit. (C) mRNA expression of Caspase-3, Bcl-2 and Bax measured by real-time PCR. (D) Up-regulation of Bcl-2 expression and down-regulation of Bax expression caused by pORF5 (data representing three independent experiments). Note: Bar graphs represent the expression of p17 Caspase-3 subunit from three independent experiments. *p < .01, **p < .05 and #p > .05.
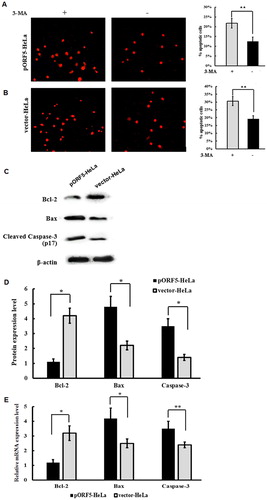
Inhibition of autophagy by 3-MA promotes apoptosis
As mentioned above, we have confirmed that pORF5 not only increased autophagy but also inhibited apoptosis. In light of the relationship between autophagy and apoptosis, we hypothesize that autophagic mechanism is likely involved in apoptosis inhibition in pORF5-HeLa cells. To analyze the correlation between the two processes triggered by pORF5 plasmid protein, TUNEL assay was performed to detect apoptotic cells after treatment with autophagy inhibitor 3-MA under starvation condition. The data showed that inhibition of autophagy in pORF5-HeLa cells significantly promoted apoptosis when compared with the same cells without treatment with 3-MA (p < .05) (). Then, the expression of apoptosis-related proteins Caspase-3, Bax and Bcl-2 in pORF5-HeLa cells was further analyzed after treatment with 3-MA under starvation condition. The data showed that the levels of pro-apoptotic molecules Bax and cleaved Caspase-3 proteins in pORF5-HeLa cells treated with 3-MA were significantly increased (p< .05), and the expression of anti-apoptotic molecule Bcl-2 in pORF5-HeLa cells treated with 3-MA was reduced (p < .05) when compared with vector-HeLa cells (). Moreover, mRNA levels of Bax and Caspase-3 in pORF5-HeLa cells were remarkably up-regulated (p < .05), whereas mRNA expression of Bcl-2 was down- regulated (p < .05) (). These results indicated that pORF5 mediates the crosstalk between autophagy and apoptosis in HeLa cells, and the suppression of autophagosome formation by 3-MA impairs autophagy and sensitizes the cells to starvation-induced apoptosis.
Figure 5. Crosstalk between autophagy and apoptosis in HeLa cells mediated by pORF5either with or without autophagy inhibitor 3-MA under starvation condition. (A) TUNEL staining of pORF5-HeLa cells and vector-HeLa cells under a fluorescence microscope (200×). (B) Percentage of TUNEL-positive cells out of the total number of cells in pORF5-HeLa and vector-HeLa cells. (C) Expression of cleaved Caspase-3, Bax and Bcl-2 determined by Western blotting. (D) Quantification of protein bands by laser densitometry after normalization with respect to β-actin. (E) mRNA expression of Caspase-3, Bax and Bcl-2 measured with real-time PCR (data representing three independent experiments). *p < .01 and **p < .05.
Autophagy and apoptosis are closely connected and share the same set of cellular regulator proteins [Citation22–25, Citation40], such as Beclin-1, a BH3-only protein identified as a Bcl-2 family interacting protein. The interaction between Bcl-2 and Beclin-1 leads to inhibition of autophagy by interfering with the formation and activity of the autophagy promoter complex, Beclin-1/hVps34 [Citation41, Citation42]. Since Bcl-2 also binds to other BH3 domain-containing proteins such as Bax, altered interaction between Beclin-1 and Bcl-2 may affect Bax function, which is essential for caspase-3 activation. As expected, our result showed that autophagy inhibitor reduced Bcl-2 expression, increased the levels of Bax and cleaved Caspase-3 proteins. This suggests that the reduced Bcl-2 expression caused by autophagy inhibition attenuates interaction between Bcl-2 and Bax and allows Bax to take an active conformation, which leads to Caspase-3 activation and apoptosis stimulation. Therefore, pORF5 coordinately regulates autophagy and apoptosis probably by elevating Beclin-1 and consequently modulating a four-way interaction among Bcl-2 proteins, Beclin-1, Bax and Caspase-3.
Conclusions
pORF5 not only could strongly induce autophagy in HeLa cells under serum starvation condition, but also inhibit apoptosis induced by TNF-α. Inhibition of autophagy promoted apoptosis in pORF5-HeLa cells which might be related to the up-regulation of Bax/Bcl-2 ratio and caspase-3 activity. These data indicated that pORF5 mediated the crosstalk between autophagy and apoptosis in HeLa cells, but further studies on the effects of autophagy induction and apoptosis inhibition trigged by pORF5 on chlamydial infection are still needed. Overall, our findings provide new insights into the role of pORF5 in autophagy and apoptosis and suggest that manipulation of host autophagy and apoptosis response can be a new approach for interventions for the prevention of C. trachomatis infection.
Author contributions
Q. H. and Z. L. collaborated to the design of the study, B. H. and Y. Z. were responsible for indirect immunofluorescence and Western blotting experiments, X. Y. and Y. W. were responsible for TUNEL experiment, S. S., H. C. and Z. Z. analyzed the data. All authors collaborated to interpret results and develop the manuscript.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Written informed consents for publication of any associated data and accompanying images were obtained from all patients or their parents, guardians or next of kin.
Ethical approval and consent to participate
All procedures performed in the present study were approved by the Ethics Committee of University of South China.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by the National Natural Science Foundation of China under grants number 81772210 and 31470277, Construct Program of the Key Discipline in Hunan Province under grant number 2011-76, Hunan Provincial Key Laboratory for Special Pathogens Prevention and Control under grant number 2014-5 and Hunan Province Cooperative Innovation Center for Molecular Target New Drug Study under grant number 2016-429.
References
- Rowhani-Rahbar A, Niccolai LM, Dunne DW, et al. Comparative epidemiology of Chlamydia trachomatis infection among men attending sexually transmitted disease clinics with and without indication for testing. Int J STD AIDS. 2006;17:453–458.
- Gerard HC, Carter JD, Hudson AP. Chlamydia trachomatis is present and metabolically active during the remitting phase in synovial tissues from patients with chronic Chlamydia-induced reactive arthritis. Am J Med Sci. 2013;346:22–25.
- Pinto CN, Dorn LD, Chinchilli VM, et al. Rural counties chlamydia and gonorrhea rates in Pennsylvania among adolescents and young adults. Ann Epidemiol. 2017;27:606–610.
- Madeleine MM, Anttila T, Schwartz SM, et al. Risk of cervical cancer associated with Chlamydia trachomatis antibodies by histology, HPV type and HPV cofactors. Int J Cancer. 2007;120:650–655.
- Jenness SM, Weiss KM, Goodreau SM, et al. Incidence of Gonorrhea and Chlamydia following human immunodeficiency virus preexposure prophylaxis among men who have sex with men: a modeling study. Clin Infect Dis. 2017;65:712–718.
- Patton DL, Sweeney YC, Baldessari AE, et al. The Chlamydia trachomatis plasmid and CT135 virulence factors are not essential for genital tract infection or pathology in female pig-tailed macaques. Infect Immun. 2018;86:1–13.
- Kari L, Whitmire WM, Olivares-Zavaleta N, et al. A live-attenuated chlamydial vaccine protects against trachoma in nonhuman primates. J Exp Med. 2011;208:2217–2223.
- Qu Y, Frazer LC, O'Connell CM, et al. Comparable genital tract infection, pathology, and immunity in rhesus macaques inoculated with wild-type or plasmid-deficient Chlamydia trachomatis serovar D. Infect Immun. 2015;83:4056–4067.
- Sigar IM, Schripsema JH, Wang Y, et al. Plasmid deficiency in urogenital isolates of Chlamydia trachomatis reduces infectivity and virulence in a mouse model. Pathogens Dis. 2014;70:61–69.
- Zhong G. Chlamydial Plasmid-Dependent Pathogenicity. Trends Microbiol. 2017;25:141–152.
- Li Z, Chen D, Zhong Y, et al. The chlamydial plasmid-encoded protein pgp3 is secreted into the cytosol of Chlamydia-infected cells. Infect Immun. 2008;76:3415–3428.
- Cao W, Zou Y, Su S, et al. Chlamydial plasmid-encoded protein pORF5 induces production of IL-1beta and IL-18 via NALP3 inflammasome activation and p38 MAPK pathway. Int J Clin Exp Med. 2015;8:20368–20379.
- Zhou H, Huang Q, Li Z, et al. PORF5 plasmid protein of Chlamydia trachomatis induces MAPK-mediated pro-inflammatory cytokines via TLR2 activation in THP-1 cells. Sci China Life Sci. 2013;56:460–466.
- Hou S, Sun X, Dong X, et al. Chlamydial plasmid-encoded virulence factor Pgp3 interacts with human cathelicidin peptide LL-37 to modulate immune response. Microbes Infect. 2019;21:50–55.
- Hou S, Dong X, Yang Z, et al. Chlamydial plasmid-encoded virulence factor Pgp3 neutralizes the antichlamydial activity of human cathelicidin LL-37. Infect Immun. 2015;83:4701–4709.
- Singh V, Finke-Isami J, Hopper-Chidlaw AC, et al. Salmonella co-opts host cell chaperone-mediated autophagy for intracellular growth. J Biol Chem. 2017;292:1847–1864.
- Chakraborty S, Castranova V, Perez MK, et al. Nanoparticles increase human bronchial epithelial cell susceptibility to respiratory syncytial virus infection via nerve growth factor-induced autophagy. Physiol Rep. 2017;5:e13344. [cited 2019 Jun 30] [14 p.].
- Jung KI, Pyo CW, Choi SY. Influenza A virus-induced autophagy contributes to enhancement of virus infectivity by SOD1 downregulation in alveolar epithelial cells. Biochem Biophys Res Commun. 2018;498:960–966.
- Kang Y, Yuan R, Xiang B, et al. Newcastle disease virus-induced autophagy mediates antiapoptotic signaling responses in vitro and in vivo. Oncotarget. 2017;8:73981–73993.
- Zhang L, Qin Y, Chen M. Viral strategies for triggering and manipulating mitophagy. Autophagy. 2018;14:1665–1673.
- Al-Younes HM, Brinkmann V, Meyer TF. Interaction of Chlamydia trachomatis serovar L2 with the host autophagic pathway. Infect Immun. 2004;72:4751–4762.
- Chen Q, Kang J, Fu C. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduct Target Ther. 2018;3:18.[cited 2019 Jun 30]
- Cooper KF. Till death do us part: the marriage of autophagy and apoptosis. Oxid Med Cell Longev. 2018;2018:1. [cited 2019 Jun 30];2018:4701275.
- Kasprowska-Liskiewicz D. The cell on the edge of life and death: crosstalk between autophagy and apoptosis. Postepy Hig Med Dosw (Online). 2017;71:825–841.
- Booth LA, Tavallai S, Hamed HA, et al. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell Signal. 2014;26:549–555.
- Cho DH, Jo YK, Hwang JJ, et al. Caspase-mediated cleavage of ATG6/Beclin-1 links apoptosis to autophagy in HeLa cells. Cancer lett. 2009;274:95–100.
- Tsapras P, Nezis IP. Caspase involvement in autophagy. Cell Death Differ. 2017;24:1369–1379.
- Ojha R, Ishaq M, Singh SK. Caspase-mediated crosstalk between autophagy and apoptosis: mutual adjustment or matter of dominance. J Cancer Res Ther. 2015;11:514–524.
- Yang J, Yao S. JNK-Bcl-2/Bcl-xL-Bax/Bak pathway mediates the crosstalk between matrine-induced autophagy and apoptosis via interplay with Beclin 1. IJMS. 2015;16:25744–25758.
- Zhong JT, Xu Y, Yi HW, et al. The BH3 mimetic S1 induces autophagy through ER stress and disruption of Bcl-2/Beclin 1 interaction in human glioma U251 cells. Cancer Lett. 2012;323:180–187.
- Waguia Kontchou C, Tzivelekidis T, Gentle IE, et al. Infection of epithelial cells with Chlamydia trachomatis inhibits TNF-induced apoptosis at the level of receptor internalization while leaving non-apoptotic TNF-signalling intact. Cell Microbiol. 2016;18:1583–1595.
- Kun D, Xiang-Lin C, Ming Z, et al. Chlamydia inhibit host cell apoptosis by inducing Bag-1 via the MAPK/ERK survival pathway. Apoptosis. 2013; 18:1083–1092.
- Igietseme JU, Omosun Y, Partin J, et al. Prevention of Chlamydia-induced infertility by inhibition of local caspase activity. J Infect Dis. 2013;207:1095–1104.
- Al-Zeer MA, Xavier A, Abu Lubad M, et al. Chlamydia trachomatis prevents apoptosis via activation of pdpk1-myc and enhanced mitochondrial binding of Hexokinase II. EBioMedicine. 2017;23:100–110.
- Zou Y, Dai W, Lei W, et al. Identification of proteins interacting with pORF5 in the pathogenesis of C. trachomatis. Am J Transl Res. 2018;10:1633–1647.
- Asanuma K, Tanida I, Shirato I, et al. MAP-LC3, a promising autophagosomal marker, is processed during the differentiation and recovery of podocytes from PAN nephrosis. FASEB J. 2003;17:1165–1167.
- Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy. 2007;3:323–328.
- Rajalingam K, Sharma M, Lohmann C, et al. Mcl-1 is a key regulator of apoptosis resistance in Chlamydia trachomatis-infected cells. PLoS One. 2008;3:e3102. [cited 2019 Jun 30] [11 p.].
- Bastidas RJ, Elwell CA, Engel JN, et al. Chlamydial intracellular survival strategies. Cold Spring Harb Perspect Med. 2013;3:a010256. [cited 2019 Jun 30] [20p.].
- Maiuri MC, Zalckvar E, Kimchi A, et al. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752.
- Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939.
- Maejima Y, Kyoi S, Zhai P, et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med. 2013;19:1478–1488.