
Abstract
Myocardial ischemia-reperfusion injury (IRI) is a severe trauma which is characterized by inflammatory reaction, oxidative stress and cardiomyocyte apoptosis. Anesthetics such as sevoflurane have been proved to exhibit cardioprotective effect on IRI and the present study aimed to explore the underlying mechanism. In this study, H9C2 cells were randomly divided into the following groups: Control group; hypoxia/reoxygenation (H/R) group; 2.5% sevoflurane (Sev) 1 h group (H9C2 cells were exposed to 1 h of 2.5% sevoflurane 24 h before H/R); 2.5% Sev 2 h group (H9C2 cells were exposed to 2 h of 2.5% sevoflurane 24 h before H/R); 2.5% sevoflurane (Sev) 2 h + LY294002 group (H9C2 cells were pretreated with 10 μL LY294002 for 24 h before sevoflurane treatment). Cell proliferation and apoptosis were examined by CCK8 assay and Flow cytometry. Then, the expression levels of key proteins, including Bcl-2, Mcl-1, iNOS, p-AKT, t-AKT, PIM1, P-Bad, p-GSK3β, t-GSK3β and cyclinD1, were examined by western blot. Furthermore, nitric oxide (NO) concentration was detected with an ELISA kit, and TNF-α, IL-1β, IL-6 and IL-10 levels were examined by western blot. The CCK8 assay and flow cytometry results indicated that sevoflurane pretreatment reduced the apoptosis of H9C2 cells with H/R injury. In addition, sevoflurane pretreatment significantly inhibited the inflammatory injury induced by H/R. Furthermore, sevoflurane activated AKT/Pim-1 and AKT/GSK3β signaling pathway. These beneficial effects of sevoflurane were canceled by phosphoinositide-3-kinase inhibitor LY294002. In conclusion, these results verified that sevoflurane attenuates H/R-induced cardiomyocyte injury via AKT/Pim-1 and AKT/GSK3β signaling pathways.
Introduction
Ischemic heart disease (IHD) is one of the most important diseases causing death worldwide [Citation1]. Currently, the clinical therapeutic application for IHD is reperfusion of the occluded artery. Off-pump coronary artery bypass (OPCAB) surgery has been used in a more widespread way [Citation2]. Ischemic myocardial damage accompanying coronary artery bypass graft surgery and subsequent reperfusion of the heart remains a clinical challenge. The evolution of the techniques and knowledge of beating heart surgery has led anesthesia toward the development of new procedures and innovations to promote patient safety and ensure high standards of care [Citation3]. Volatile anesthetic agents have the potential to provide myocardial protection by anesthetic preconditioning [Citation4]. The volatile anesthetic sevoflurane is typically used in general anesthesia for patients undergoing surgery, including heart surgery [Citation5, Citation6]. However, the precise mechanism of sevoflurane’s role in cardio-protection has not been elucidated.
AKT is a nodal signaling kinase which is known to be a crucial signaling protein in the myocardium [Citation7]. Previous evidence indicates that AKT action is actually mediated by a downstream kinase called Pim-1 [For review see [Citation4]]. Pim-1 is a serine-threonine kinase, one of a three-member family of serine/threonine kinases belonging to the calmodulin-dependent protein kinase-related group [Citation8]. Pim-1 was originally discovered as the proviral integration site for the Moloney murine leukemia virus [Citation9] and originally identified as a cellular oncogene that could inhibit apoptosis and promote proliferation [Citation10]. Pim-1 inhibited cardiomyocyte apoptosis with concomitant increases in Bcl-2 and Bcl-X (L) protein levels, as well as in Bad phosphorylation levels [Citation11]. Glycogen synthase kinase-3β (GSK3β) is a Ser/Thr protein kinase which has emerged as a key end-effector of multiple signaling pathways for cardio-protection [Citation12]. The evidence showed that cyclin D1 is one of the target genes that may be activated by β-catenin for cell proliferation. Meanwhile, cyclin D1 can promote cell cycle progression [Citation13]. The AKT/GSK3β/cyclin D1 pathway plays a key role in regulating cell apoptosis [Citation14].
Therefore, the present study aimed to investigate the cardioprotective effect of sevoflurane and the underlying protection mechanism focusing on AKT/Pim-1 and AKT/GSK3β pathways.
Materials and methods
Cell culture
H9C2 cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in Dulbecco's modified Eagle's medium, Low Glucose (DMEM/FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), supplemented with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA, USA) and 100 units/mL penicillin and 100 μg/mL streptomycin (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) at standard culture conditions (37 °C, 5% CO2 and 95% humidity). Cells were subjected to experimental procedures at 80–90% confluence.
H/R injury model in vitro
H/R injury model in vitro was established as previously described [Citation15]. Briefly, H9C2 cells were cultured in serum-free and low-glucose DMEM in a hypoxia chamber with atmosphere containing 95% N2 and 5% CO2 for 6 h at 37 °C, and then the culture medium was replaced with fresh medium containing 10% FBS, after which the cells were cultured in a gas mixture of 95% O2 and 5% CO2 for another 6 h at 37 °C. H9C2 cells were divided into four experimental groups: Control, model (hypoxia/reoxygenation, H/R), 2.5% Sev 1 h (H9C2 cells were exposed to 1 h of 2.5% sevoflurane 24 h before H/R), 2.5% Sev 2 h (H9C2 cells were exposed to 2 h of 2.5% sevoflurane 24 h before H/R). For the purpose of sevoflurane treatment, H9C2 cells were seeded onto plates and cultured overnight. Then cell plates were placed in a sterile sealed container connected to an anesthetic vaporizer for 1 h or 2 h. The concentrations of sevoflurane were monitored by a gas monitor. The 2.5% concentration of sevoflurane was chosen based on a previous study [Citation16]. Meanwhile, phosphoinositide-3-kinase inhibitor LY294002 was also applied to investigate the potential molecular mechanism in the beneficial effects of sevoflurane.
Cell counting kit-8 (CCK-8) assay
Cell viability was determined by Cell Counting Kit-8 (CCK-8, Dojinodo, Shanghai, China) according to the manufacturer’s instructions. In brief, H9C2 cells (3 × 103 cells/well) were cultured in 96-well plates for 24 h. After the indicated treatments, cells were incubated with 10 μL CCK-8 solution for 1 h. The optical density (OD) value at 450 nm was measured by a microplate reader (Dynex, Chantilly, VA, USA).
Flow cytometry
Cellular apoptosis was evaluated using the Annexin V-FITC Apoptosis Detection Kit (Beyotime) according to the manufacturer’s instructions. Briefly, the treated H9C2 cells (1 × 105) were harvested and re-suspended in 200 μL binding buffer. Then, the cells were double-stained with 10 µL fluorescein (FITC)-conjugated Annexin V and 5 µL propidium iodide (FITC-Annexin V/PI) (BD Biosciences, San Diego, CA, USA) for 15 min at room temperature in the dark. The apoptosis rates were analyzed on a flow cytometer (FACSCalibur; BD Biosciences).
Enzyme-linked immunosorbent assay (ELISA)
H9C2 cells were cultured in six plates. After the indicated treatment, the levels of NO in H9C2 cells were measured using the NO assay Kit (SBJ-R0010, Nanjing SenBeiJia Biological Technology Co., Ltd.) in accordance with the manufacturer’s protocol. NO was considered as a reactive nitrogen species and the NO levels were used as a marker of oxidative stress.
Western blot analysis
Cells were harvested and lysed with 0.5 mL radioimmunoprecipitation assay buffer (Beyotime Institute of Biotechnology, Hai men, China). The protein concentration was determined with a BCA Protein Assay kit (P0010; Beyotime Institute of Biotechnology) according to the manufacturer's protocol. Protein samples (10 μg per lane) from the lysates were separated by 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes. Subsequently, the membranes were blocked with 5% skimmed milk for about 2 h at room temperature and incubated with primary antibodies: Anti-Bcl-2 (1:1000; SAB4500003; Sigma-Aldrich; Merck KGaA), Anti-Mcl1(1:1000; SAB4501844; Sigma-Aldrich; Merck KGaA) , Anti-TNF-α (1:1000; ab6671; Abcam), Anti-IL-1β (1:1000; BS6067; Bioworld), Anti-IL-6 (1:1000; BS6419; Bioworld), Anti-IL-10 (1:1000; ab9969; Abcam), Anti-iNOS (1:2000; ab15323; Abcam), Anti-p-AKT (1:1000; SAB4301414; Sigma-Aldrich; Merck KGaA), Anti-t-AKT (1:1000; SAB4500796; Sigma-Aldrich; Merck KGaA), Anti- PIM1 (1:1000; BS2988; Bioworld), Anti-p-Bad (1:1000; SAB4301255; Sigma-Aldrich; Merck KGaA), Anti-Bad (1:1000; SAB4300342; Sigma-Aldrich; Merck KGaA), Anti-p-GSK3β (pSer9) (1:1000; SAB4300001; Sigma-Aldrich; Merck KGaA), GSK-3β (1:1000; MAB2506; R&D Systems), Anti-Cyclin D1 (1:10000; ab134175; Abcam) and Anti-GAPDH (1:20000; G8795; Sigma-Aldrich; Merck KGaA) at 4˚C overnight. Thereafter, the PVDF membranes were treated with horseradish peroxidase (HRP)-conjugated secondary antibody (Anti-Mouse IgG; 1:10000; A9917; Sigma-Aldrich; Merck KGaA) at 37 °C for 1 h. Protein bands were visualized by an enhanced chemiluminescence kit (sc-2048, Sigma-Aldrich, Merck KGaA). Data were analyzed with Image Pro Plus v.6.0 software (Media Cybernetics, Inc., Rockville, MD, USA) for densitometry. GAPDH (glyceraldehyde 3-phosphate dehydrogenase) was used as an endogenous control.
Statistical analysis
SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA) was used for statistical analysis. All the experiments were performed independently at least three times. Data are presented as mean values with standard error of the means (±SEM). Statistical significance was determined by Student's t-test or one-way analysis of variance (ANOVA) followed by the Scheffe post-hoc test. Differences were considered to be statistically significant at a probability level of p < 0.05.
Results and discussion
Sevoflurane protected H9C2 cells from H/R-induced decrease of cell viability
To analyze the protective effect of sevoflurane on H/R-induced injury in H9C2 cells, we first detected the cell viability by CCK8 assay following treatment with 2.5% sevoflurane for 1 h or 2 h. As shown in , cell viability was significantly reduced in H/R-treated H9C2 cells, and pretreatment with 2.5% sevoflurane for 1 h and 2 h significantly attenuated H/R-induced reduction of cell viability in H9C2 cells.
Figure 1. Sevoflurane protected H9C2 cells from H/R-induced decrease of cell viability. H9c2 cells were exposed to 1 h and 2 h of 2.5% sevoflurane 24 h before H/R for 24 h followed by H/R. Cell viability was measured with CCK8 assay. Data are represented as mean values ± SEM. ***p < 0.001 vs. control group, #p < 0.05, ##p < 0.01 vs. H/R group. H/R, hypoxia/reoxygenation; Sev, sevoflurane.
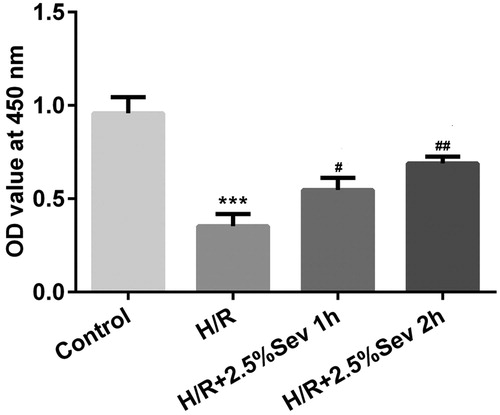
Sevoflurane inhibited H9C2 cells apoptosis induced by H/R injury
To further elucidate the anti-apoptotic role of sevoflurane in H/R-treated H9C2 cells, cell apoptosis was detected by flow cytometry. As shown in , the percentage of apoptotic cells was significantly increased after H/R treatment, whereas pretreatment with 2.5% sevoflurane for 1 h and 2 h significantly reduced the apoptotic ratio induced by H/R. In addition, the expression levels of apoptosis-associated proteins such as Bcl-2 and Mcl1 were further assessed by western blot analysis. As shown in , the levels of Bcl-2 and Mcl1 were significantly downregulated in H/R-treated H9C2 cells, whereas sevoflurane significantly upregulated H/R-induced downregulation of Bcl-2 and Mcl1 expression in H9C2 cells.
Figure 2. Sevoflurane inhibited H9C2 cells from H/R-induced apoptosis. H9C2 cells were exposed to 1 h and 2 h of 2.5% sevoflurane for 24 h followed by H/R. (A) The apoptotic ratio of H9C2 cells was detected by flow cytometry. (B) The protein levels of Bcl-2 and Mcl1 were detected by western blot. Data are represented as means ± SEM. ***p < 0.001 vs. control group, #p < 0.05, ##p < 0.01, ###p < 0.001 vs. H/R group. H/R, hypoxia/reoxygenation; Sev, sevoflurane.
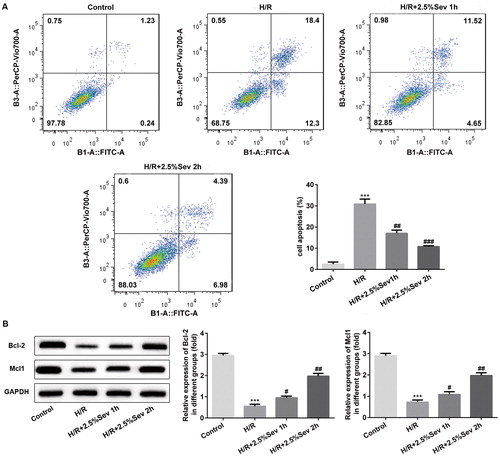
Sevoflurane inhibited inflammation of H9C2 cells induced by H/R injury
To measure the effect of sevoflurane on the H/R-induced inflammation in H9C2 cells, we detected TNF-α, IL-1β, IL-6 and IL-10 levels by western blot. Compared with the control group, the levels of TNF-α, IL-1β and IL-6 were significantly upregulated and IL-10 was significantly downregulated in H/R-treated H9C2 cells. As shown in , sevoflurane pretreatment for 1 h and 2 h significantly decreased the levels of TNF-α, IL-1β and IL-6 and increased the IL-10 level compared with the H/R group.
Figure 3. Sevoflurane inhibited inflammation of H9C2 cells induced by H/R injury. The protein levels of TNF-α, IL-1β, IL-6 and IL-10 were detected by western blot. GAPDH served as an internal loading control. Data are represented as means ± SEM. ***p < 0.001 vs. control group, #p < 0.05, ##p < 0.01, ###p < 0.001 vs. H/R group. H/R, hypoxia/reoxygenation; Sev, sevoflurane.
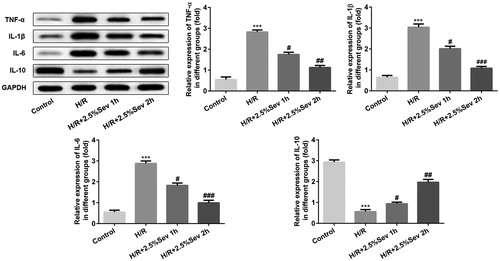
Sevoflurane mitigated H/R-induced NO production
Previous studies have shown that NO improves H/R-induced cardiac cell damage through functional and pathophysiological changes [Citation17]. Endothelial NOS (eNOS) is a downstream enzyme of AKT responsible for the production of NO, while inducible NOS (iNOS) produces NO at high concentrations [Citation18]. To confirm the effect of sevoflurane on NO production in H/R-treated H9C2 cells, we examined the NO level by ELISA assay and the protein level of iNOS by western blot. We found that H/R increased the level of NO and the expression of iNOS, but these effects were correspondingly reversed by sevoflurane pretreatment ().
Figure 4. Sevoflurane mitigated H/R-induced increase of NO production. (A) The level of NO was measured by ELISA assay. (B) The protein level of iNOS was measured by western blot. GAPDH served as an internal loading control. Data are represented as means ± SEM. ***p < 0.001 vs. control group, #p < 0.05, ##p < 0.01 vs. H/R group. H/R, hypoxia/reoxygenation; Sev, sevoflurane.
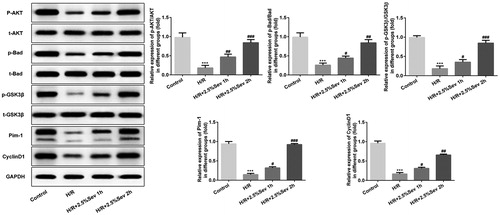
Sevoflurane activated AKT/pim-1 and AKT/GSK3β signaling pathway
To explore the underlying cardioprotection mechanism of sevoflurane, we detected some crucial proteins related to AKT/Pim-1 and AKT/GSK3β signaling pathway, such as p-AKT, t-AKT, PIM1, p-Bad, t-Bad, p-GSK3β, t-GSK3β and cyclin D1. Compared with the control group, H/R injury significantly decreased the levels of p-AKT, Pim-1, P-Bad, p-GSK3β and cyclin D1, which were increased in the sevoflurane pretreatment group ().
Figure 5. Sevoflurane activated AKT/Pim-1 and AKT/GSK3β signaling pathway. The protein levels of p-AKT, t-AKT, PIM1, P-Bad, p-GSK3β, t-GSK3β and cyclin D1 were detected by western blot. GAPDH served as an internal loading control. Data are represented as means ± SEM. ***p < 0.001 vs. control group, #p < 0.05, ##p < 0.01, ###p < 0.001 vs. H/R group. H/R, hypoxia/reoxygenation; Sev, sevoflurane.
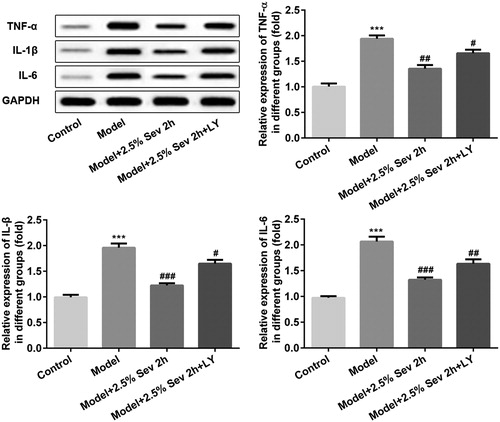
Sevoflurane reversed inflammation induced by H/R injury via AKT/pim-1 and AKT/GSK3β signaling pathway in vitro
To measure whether sevoflurane influenced inflammation via AKT/Pim-1 and AKT/GSK3β signaling pathway, H9C2 cells were pretreated with 10 μL LY294002 for 24 h before 2.5% sevoflurane treatment. TNF-α, IL-1β and IL-6 levels were detected by western blot. As shown in , the levels of TNF-α, IL-1β and IL-6 were significantly upregulated in H/R-treated H9C2 cells compared with the control group. Sevoflurane significantly decreased the levels of TNF-α, IL-1β and IL-6 expression compared with the H/R group, which were correspondingly reversed by LY294002 pretreatment.
Figure 6. Sevoflurane inhibited H/R-induced inflammation in H9C2 cells via AKT/Pim-1 and AKT/GSK3β signaling pathway. In 2.5% sevoflurane (Sev) 2 h group, H9C2 cells were exposed to 2 h of 2.5% sevoflurane 24 h before H/R treatment; 2.5% sevoflurane (Sev) 2 h + LY294002 group, H9C2 cells were pretreated with 10 μL LY294002 for 24 h before sevoflurane treatment. The protein levels of TNF-α, IL-1β, and IL-6 were detected by western blot. GAPDH served as an internal loading control. Data are represented as means ± SEM. ***p < 0.001 vs. control group, #p < 0.05, ##p < 0.01, ###p < 0.001 vs. H/R group. H/R, hypoxia/reoxygenation; Sev, sevoflurane; LY, phosphoinositide-3-kinase inhibitor LY294002.
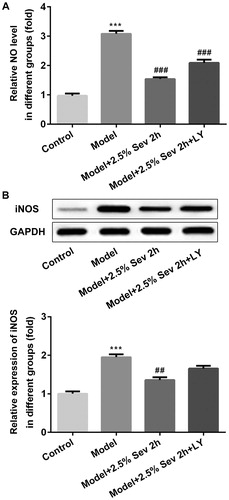
Sevoflurane mitigated H/R-Induced NO production via AKT/pim-1 and AKT/GSK3β signaling pathway
To confirm whether sevoflurane inhibited NO and iNOS production in H/R-treated H9C2 cells via AKT/Pim-1 and AKT/GSK3β signaling pathway, we examined the NO level by ELISA assay and the protein expression of iNOS by western blot. As shown in , H/R increased the level of NO and the expression of iNOS compared with the control group. Sevoflurane significantly decreased the levels of NO and iNOS compared with the H/R group, which were correspondingly reversed by LY294002 pretreatment.
Figure 7. Sevoflurane mitigated H/R-Induced NO production via AKT/Pim-1 and AKT/GSK3β signaling pathway. In 2.5% sevoflurane (Sev) 2 h group, H9C2 cells were exposed to 2 h of 2.5% sevoflurane 24 h before H/R treatment; 2.5% sevoflurane (Sev) 2 h + LY294002 group, H9C2 cells were pretreated with 10 μL LY294002 for 24 h before sevoflurane treatment. (A) The level of NO was measured by ELISA assay. (B) The protein level of iNOS was measured by western blot. GAPDH served as an internal loading control. Data were represented as means ± SEM. ***p < 0.001 vs. control group, ##p < 0.01, ###p < 0.001 vs. H/R group. H/R, hypoxia/reoxygenation; Sev, sevoflurane; LY, phosphoinositide-3-kinase inhibitor LY294002.
Sevoflurane inhibited H9C2 cells apoptosis induced by H/R injury via AKT/pim-1 and AKT/GSK3β signaling pathway
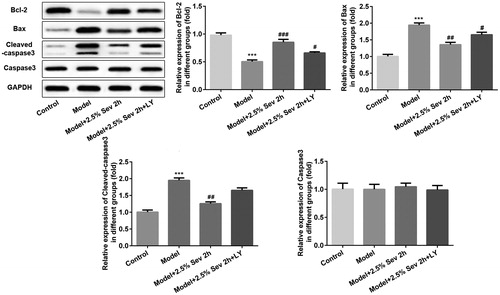
To further elucidate the anti-apoptotic role of sevoflurane in H/R-treated H9C2 cells via AKT/Pim-1 and AKT/GSK3β signaling pathway, cell apoptosis was detected by flow cytometry and the expression levels of apoptosis-associated proteins such as Bcl-2, Bax, cleaved capase3 and capase3 were further assessed by western blot analysis. As shown in , the percentage of apoptotic cells was significantly increased after H/R treatment, and pretreatment with 2.5% sevoflurane for 2 h significantly reduced the apoptotic ratio induced by H/R. In addition, the anti-apoptotic role of sevoflurane was reversed by LY294002 pretreatment. As shown in , the level of Bcl-2 was significantly downregulated and the levels of Bax and cleaved capased3 were significantly upregulated in H/R-treated H9C2 cells, whereas sevoflurane significantly upregulated H/R-induced downregulation of Bcl-2 expression and downregulated H/R-induced upregulation of Bax and cleaved capased3 levels in H9C2 cells. Pretreatment with 10 μL LY294002 for 24 h correspondingly reversed the anti-apoptotic effect of sevoflurane in H/R-treated H9C2 cells.
Figure 8. Sevoflurane inhibited H9C2 cells from H/R-induced apoptosis via AKT/Pim-1 and AKT/GSK3β signaling pathway. In 2.5% sevoflurane (Sev) 2 h group, H9C2 cells were exposed to 2 h of 2.5% sevoflurane 24 h before H/R treatment; 2.5% sevoflurane (Sev) 2 h + LY294002 group, H9C2 cells were pretreated with 10 μL LY294002 for 24 h before sevoflurane treatment. (A) The apoptotic ratio of H9C2 cells was detected by flow cytometry. (B) Quantitative analysis of apoptotic rate. Data are represented as means ± SEM. ***p < 0.001 vs. control group, ##p < 0.01 vs. H/R group. H/R, hypoxia/reoxygenation; Sev, sevoflurane; LY, phosphoinositide-3-kinase inhibitor LY294002.
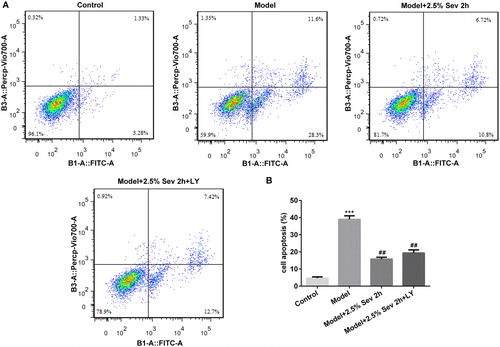
Figure 9. Sevoflurane inhibited H9C2 cells from H/R-induced apoptosis via AKT/Pim-1 and AKT/GSK3β signaling pathway. In 2.5% sevoflurane (Sev) 2 h group, H9C2 cells were exposed to 2 h of 2.5% sevoflurane 24 h before H/R treatment; 2.5% sevoflurane (Sev) 2 h + LY294002 group, H9C2 cells were pretreated with 10 μL LY294002 for 24 h before sevoflurane treatment. The expression levels of apoptosis-associated proteins such as Bcl-2, Bax, cleaved capase3 and capase3 were further assessed by western blot analysis. GAPDH served as an internal loading control. Data are represented as means ± SEM. ***p < 0.001 vs. control group, #p < 0.05, ##p < 0.01, ###p < 0.001 vs. H/R group. H/R, hypoxia/reoxygenation; Sev, sevoflurane; LY, phosphoinositide-3-kinase inhibitor LY294002.
Taken together, our results revealed that sevoflurane effectively protected the cardiomyocytes against H/R injury, as evidenced by mitigating cell apoptosis and inflammation injury in H/R-treated H9C2 cells. Furthermore, we demonstrated that the mechanism attributable to the cardioprotective effect of sevoflurane may be the activation of AKT/Pim-1 and AKT/GSK3β signaling pathway.
Sevoflurane is a commonly used anesthetic with few clinical side effects [reviewed in [Citation19]]. Previous studies have demonstrated that sevoflurane treatment could show effective myocardial and cardioprotection [Citation20, Citation21]. Moreover, preconditioning can inhibit cardiomyocyte apoptosis and alleviate myocardial inflammation [Citation22, Citation23], but the potential mechanism remains unclear. The results in our present study were in accordance with previous studies. We found that sevoflurane could protect H9c2 cells from H/R-induced cell injury, such as cell apoptosis, inflammation and oxidative stress.
Some reports showed that AKT activation shows pro-survival and proliferative effects in the myocardium [Citation24, Citation25]. Inactivation of AKT may account for increased cardiomyocyte apoptosis because AKT directly controls the phosphorylation and sequestration of the Bad [Citation26]. Cardiac actions of AKT are mainly ascribed to Pim-1 dependence. AKT activation induced Pim-1 expression, whereas forced expression of AKT failed to protect the infarcted myocardium in Pim-1-deficient mice [Citation11]. The results in our present study revealed that sevoflurane could display its cardioprotective effect via AKT/Pim-1 and AKT/GSK3β signaling pathway.
Conclusions
The obtained results provided further evidence of the cardioprotective role of sevoflurane, which could reduce H/R-induced cardiomyocyte injury in vitro. The potential mechanism may be closely related to the activation of AKT/Pim-1 and AKT/GSK3β signaling pathway.
Authors’ contributions
WS Zhao, TS Zhang, LL Xu, Y Yang, YC Wang, ZN Jiang searched the literature, designed the study and performed the experiments. WS Zhao, TS Zhang, LL Xu, Y Yang, YC Wang, ZN Jiang analyzed data and wrote the manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This study was supported by Hangzhou Municipal Science and Technology Commission (No. 20170533B63) and Zhejiang Provincial Science Technology Foundation of China (No. 2017C37075 and No. 2018C37117).
Availability of data and materials
The analyzed data sets generated during the present study are available from the corresponding author upon reasonable request.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
References
- Ren L, Wang Q, Chen Y, et al. Involvement of microRNA-133a in the protective effect of hydrogen sulfide against ischemia/reperfusion-induced endoplasmic reticulum stress and cardiomyocyte apoptosis. Pharmacology. 2019;103(1-2):1–9.
- Nakano J, Okabayashi H, Noma H, et al. The impact of incomplete revascularization and angiographic patency on midterm results after off-pump coronary artery bypass grafting. J Thorac Cardiovasc Surg. 2014;147(4):1225–1232.
- Hemmerling TM, Romano G, Terrasini N, et al. Anesthesia for off-pump coronary artery bypass surgery. Ann Card Anaesth. 2013;16(1):28–39.
- Kunst G, Klein AA. Peri-operative anaesthetic myocardial preconditioning and protection–cellular mechanisms and clinical relevance in cardiac anaesthesia. Anaesthesia. 2015;70(4):467–482.
- Yang XL, Wang D, Zhang GY, et al. Comparison of the myocardial protective effect of sevoflurane versus propofol in patients undergoing heart valve replacement surgery with cardiopulmonary bypass. BMC Anesthesiol. 2017;17(1):37.
- Zhang Y, Lin W, Shen S, et al. Randomized comparison of sevoflurane versus propofol-remifentanil on the cardioprotective effects in elderly patients with coronary heart disease. BMC Anesthesiol. 2017;17(1):104. DOI:
- Song HP, Chu ZG, Zhang DX, et al. PI3K-AKT pathway protects cardiomyocytes against hypoxia-induced apoptosis by mitoKATP-mediated mitochondrial translocation of pAKT. Cell Physiol Biochem. 2018;49(2):717–727.
- Bullock AN, Debreczeni J, Amos AL, et al. Structure and substrate specificity of the Pim-1 kinase. J Biol Chem. 2005;280(50):41675–41682.
- Tursynbay Y, Zhang J, Li Z, et al. Pim-1 kinase as cancer drug target: an update. Biomed Rep. 2016;4(2):140–146.
- Aweya JJ, Wang W, Zhang Y, et al. Identification and molecular characterization of the Pim1 serine/threonine kinase homolog in Litopenaeus vannamei. Fish Shellfish Immunol. 2018;74:491–500.
- Muraski JA, Rota M, Misao Y, et al. Pim-1 regulates cardiomyocyte survival downstream of Akt. Nat Med. 2007;13(12):1467–1475.
- Juhaszova M, Zorov DB, Yaniv Y, et al. Role of glycogen synthase kinase-3beta in cardioprotection. Circ Res. 2009;104(11):1240–1252.
- Takahashi-Yanaga F, Sasaguri T. GSK-3beta regulates cyclin D1 expression: a new target for chemotherapy. Cell Signal. 2008;20(4):581–589.
- Wang CD, Yuan CF, Bu YQ, et al. Fangchinoline inhibits cell proliferation via Akt/GSK-3beta/cyclin D1 signaling and induces apoptosis in MDA-MB-231 breast cancer cells. Asian Pac J Cancer Prev. 2014;15(2):769–773.
- Zhao S, Wang Y, Zhang X, et al. Melatonin protects against hypoxia/reoxygenation-induced dysfunction of human umbilical vein endothelial cells through inhibiting reactive oxygen species generation. Acta Cardiol Sin. 2018;34(5):424–431.
- Wang Z, Cui R, Wang K. Effects of sevoflurane pretreatment on the apoptosis of rat H9c2 cardiomyocytes and the expression of GRP78. Exp Ther Med. 2018; 15:2818–2823.
- Xu Y, Wang W, Jin K, et al. Perillyl alcohol protects human renal tubular epithelial cells from hypoxia/reoxygenation injury via inhibition of ROS, endoplasmic reticulum stress and activation of PI3K/Akt/eNOS pathway. Biomed Pharmacother. 2017; 95:662–669.
- Patruno A, Franceschelli S, Pesce M, et al. Novel aminobenzyl-acetamidine derivative modulate the differential regulation of NOSs in LPS induced inflammatory response: role of PI3K/Akt pathway. Biochim Biophys Acta. 2012;1820(12):2095–2104.
- Brioni JD, Varughese S, Ahmed R, et al. A clinical review of inhalation anesthesia with sevoflurane: from early research to emerging topics. J Anesth. 2017;31(5):764–778.
- Zhao J, Wang F, Zhang Y, et al. Sevoflurane preconditioning attenuates myocardial ischemia/reperfusion injury via caveolin-3-dependent cyclooxygenase-2 inhibition. Circulation. 2013;128(11_suppl_1):S121–S129.
- Lamberts RR, Onderwater G, Hamdani N, et al. Reactive oxygen species-induced stimulation of 5'AMP-activated protein kinase mediates sevoflurane-induced cardioprotection. Circulation. 2009;120(11_suppl_1):S10–S15.
- Kojima A, Kitagawa H, Omatsu-Kanbe M, et al. Sevoflurane protects ventricular myocytes against oxidative stress-induced cellular Ca2+ overload and hypercontracture. Anesthesiology. 2013;119(3):606–620.
- Singh P, Chauhan S, Jain G, et al. Comparison of cardioprotective effects of volatile anesthetics in children undergoing ventricular septal defect closure. World J Pediatr Congenit Heart Surg. 2013;4(1):24–29.
- Shiraishi I, Melendez J, Ahn Y, et al. Nuclear targeting of Akt enhances kinase activity and survival of cardiomyocytes. Circ Res. 2004;94(7):884–891.
- Yan A, Li G, Zhang X, et al. Pro-survival effect of Dock180 overexpression on rat-derived H9C2 cardiomyocytes. Med Sci Monit Basic Res. 2013;19:12–19.
- Katare RG, Caporali A, Oikawa A, et al. Vitamin B1 analog benfotiamine prevents diabetes-induced diastolic dysfunction and heart failure through Akt/Pim-1-mediated survival pathway. Circ Heart Fail. 2010;3(2):294–305.