
Abstract
Carrot (Daucus carota L.) is a versatile vegetable crop and the most economically important member of the Apiaceae family. While there are several important cultivated species in the family such as celery, parsley, cumin, fennel, coriander and parsnip, molecular genetic research in Apiaceae is relatively limited compared to other agriculturally important taxa. In the present work, an in silico approach was employed in order to develop chromosome-anchored simple sequence repeat (SSR) markers from the carrot genome assembly. A total of 55,386 markers were developed and marker loci that correspond to protein coding sequences were determined. In silico mapping analysis predicted that 51,160 of these were single-locus markers and 4,226 amplified more than one locus. Cross-species transferability of the markers was assessed using the fennel (Foeniculum vulgare Mill.) draft genome sequence, resulting in the identification of 578 low-copy transferable markers. These markers can serve for the purposes of interspecific genomic synteny studies and comparative gene identification/cloning. A subset of 50 markers was evaluated on DNA from 17 accessions of carrot. As a result, 46 (92%) produced amplicons from all genotypes, of which 28 (61%) displayed polymorphisms among the 17 carrot accessions, confirming the potential of the newly developed markers to reveal genotypic diversity in cultivated carrot. With the present work, carrot chromosomes were saturated with sequence-specific markers, which constitute a physical map of the carrot genome. The collection of markers will serve as practical molecular tools for germplasm characterization, gene tagging and molecular breeding studies in this important crop species.
Introduction
Carrot (Daucus carota L.) (2n = 2x = 18) is the most economically important member of the Apiaceae (Umbelliferae) family, and is cultivated mainly for its nutritious taproot [Citation1,Citation2]. Carotenoid-rich carrot roots constitute a major source of provitamin A in the human diet [Citation3,Citation4]. In addition to genotypes rich in Vitamin A precursor carotenoids, the carrot germplasm includes genotypes that accumulate other health-beneficial compounds such as lutein, lycopene, and anthocyanins [Citation5,Citation6]. Carrot production is increasing in parallel with growth in the market for cut-carrot products, increased recognition of carrot’s health-beneficial attributes, and development of new cultivars adapted to warmer climates [Citation7]. Indeed, carrot is among the ten most commonly cultivated vegetable crops worldwide [Citation8]. While the Apiaceae family hosts several important cultivated species including carrot, celery, parsley, fennel, cumin, coriander and parsnip, molecular genetic research in Apiaceae has lagged far behind other agronomically important families such as Solanaceae, Fabaceae, Brassicaceae and Poaceae. As a result, the genomic resources specific for carrot and other members of the Apiaceae are relatively limited [Citation2–4]. On the other hand, the first assembled genome sequence that represents nine carrot chromosomes was recently made available [Citation4] and constitutes an important resource that allows the development of molecular genetic tools in carrot.
Simple sequence repeat (SSR) markers are among the most frequently utilized marker systems in plant molecular genetics. SSRs are abundant in the genome and are often associated with low-copy, transcribed fractions of plant genomes [Citation9]. Therefore, it is frequently feasible to associate SSR alleles with phenotypes [Citation10]. The codominant nature of SSR loci allows distinguishing homozygote and heterozygote genotypes and a single SSR marker can produce a large number of alleles. Moreover, basic molecular biology lab equipment is sufficient to genotype SSRs [Citation11,Citation12]. As a result, SSR markers remain extensively utilized molecular genetic tools in plant genetics and breeding [Citation13–15].
The development of efficient computational genomics tools for sequence analyses accompanied by the continuous accumulation of assembled genome sequences in public databases made genome-wide marker development efficient and productive [Citation11,Citation14,Citation16]. Consequently, in silico high-throughput marker development approaches have almost totally replaced microsatellite-enriched library based methods. Accordingly, high-throughput development of SSR markers via mining publicly available genome assemblies has recently been reported for diverse crop plant taxa, including wheat [Citation17], chickpea [Citation18], cucumber [Citation14], peach [Citation19], melon [Citation20], bitter melon [Citation21], sesame [Citation22], hazelnut [Citation15], seven different Nicotiana species [Citation23] and 16 different tree species [Citation24].
SSR markers specific to the carrot genome are actually scarce and the first reports on the development of large numbers of SSRs are relatively recent [Citation1,Citation2]. Cavagnaro et al. [Citation2] developed 300 SSR markers in carrot using an SSR enriched genomic library and BAC-end sequences. Iorizzo et al. [Citation1] identified SSRs in a carrot transcriptome assembly from four genotypes and made available a set of 114 markers they detected as polymorphic by in silico analyses. Since then, these SSR markers have been used in the majority of carrot genomic studies. For example Baranski et al. [Citation25] determined genetic diversity and population structure of a collection of 88 carrot genotypes from diverse geographical locations. Ipek et al. [Citation26] evaluated the genetic diversity in a local purple carrot population in Turkey. Ma et al. [Citation27] investigated the molecular genetic relationships among Chinese and Western orange carrot accessions, and Mandel et al. [Citation28] deduced the patterns of gene flow between cultivated and wild forms of carrot. These available SSR loci were also utilized for mapping anthocyanin biosynthesis genes [Citation29], genes associated with flower architecture and fertility [Citation30], and QTLs for nematode resistance [Citation31]. Yet, the pool of available SSR markers is insufficient to cover the carrot genome with high resolution, and the currently available marker loci have not yet been anchored to their chromosomal locations.
Apart from SSRs, sequence-based markers developed in carrot can be exemplified with the works of Iorizzo et al. [Citation32] and Stelmach et al. [Citation33], where 4000 single nucleotide polymorphism (SNP) and 209 Intron Length Polymorphism (ILP) markers were developed, respectively. A hybridization-based genotyping strategy, DArT (Diversity Arrays Technology) genotyping, was also applied on carrot [Citation34].
The present study describes the high-throughput development of SSR markers in carrot. A chromosome-scale carrot genome assembly comprising nine scaffolds that represent the nine carrot chromosomes [Citation4] was mined for SSRs. The expected amplification profiles of the newly identified markers were also analyzed and SSR flanking sequences that correspond to protein-coding loci were determined. Mapping analysis using a scaffold-level fennel genome assembly determined markers with cross-species transferability. Further work involved laboratory evaluation of a subset of SSR markers using DNA from 17 carrot landraces, verifying the effectiveness of the marker design process and displaying the potential of the SSR loci to reveal genomic polymorphisms.
Materials and methods
Simple sequence repeat mining in the carrot genome
Genome assembly of cultivated carrot (Daucus carota subsp. sativus) was retrieved from GenBank (https://www.ncbi.nlm.nih.gov/nuccore/LNRQ00000000) [Citation4]. GMATA (Genome-wide Microsatellite Analyzing Tool Package) [Citation35] software was utilized for mining the genome assembly for SSRs and designing primers from the flanking sequences. Perl, R and Java running environments were established prior to running the GMATA software. Genome assembly scaffolds were imported to the ‘SSR identification’ module of GMATA in FASTA format. Applied microsatellite search parameters for mononucleotide repeats were: Min. length (nt): 1, Max. length (nt): 1, Min. repeat-times: 10. Search parameters for tandem repeats of two to six nucleotides were: Min. length (nt): 2, Max. length (nt): 6, Min. repeat-times: 5. SSR loci information files (.ssr) were generated that contained SSR start & end positions on chromosome sequences, SSR motifs and number of repeat units.
Marker design and mapping in the carrot and fennel genome assemblies
The ‘Marker designing’ module of GMATA was utilized in order to design primers for the identified microsatellites. The genome assembly in FASTA format and output files generated by the ‘SSR identification’ module (.ssr) were imported to the ‘Marker designing’ module as the ‘Sequence file’ and the ‘SSR loci file’, respectively. Parameters for marker design were: Min. amplicon size: 100 bp, Max. amplicon size: 300 bp, Optimal annealing Tm: 60 °C, Flanking sequence length: 400 bp, Max. template length (the genome is partitioned to individual segments for each SSR): 2000 bp. The output .seq file contained the repeat loci centered within 400 bases of flanking sequence on each side. The output files with .mk and .sts extensions were generated that contained left and right primer sequences, annealing temperatures, primer locations on chromosomes and expected amplicon sizes. The ‘e-Mapping’ module was utilized to run the e-PCR algorithm [Citation36] in order to generate amplicons and assign to them marker locations on carrot chromosomes. The module was also utilized for the genome-wide cross-species mapping of the markers on fennel genome assembly. The draft genome assembly of fennel (Foeniculum vulgare Mill.) at the scaffold level was accessed at https://www.ncbi.nlm.nih.gov/assembly/GCA_003724115.1/#/st. For the e-PCR, FASTA files that contained the carrot and fennel genome assemblies and output files generated by the ‘Marker designing’ module (.sts files) were imported as the ‘Sequence file’ and the ‘Marker file’, respectively. Max. mismatch (-n) and Max. indel (-g) parameters were set as ‘0’. The output file (.emap) provided detailed amplification patterns of the markers with calculated amplicon sizes and target positions on chromosomes, and identified single-locus and multi-locus markers.
Functional annotation of the marker loci and similarity search against existing markers
The functional annotation of SSR loci was performed by high-throughput alignment and comparison of SSR loci with the Identical Protein Groups Database of carrot (https://www.ncbi.nlm.nih.gov/ipg/?term=daucus%20carota), which includes peptide sequences merged based on protein identity. For this purpose, genomic sequences that harbor the simple sequence repeats (query sequences) were converted to FASTA format and the Identical Protein Groups Database was retrieved as a single FASTA file. The query sequence for each SSR marker was the repeat locus with 400 bases of flanking sequence on each side. Thus, each query consisted of at least 810 bases [800 bases of flanking sequence + the minimum SSR size (Min-length (nt): 2, Min. repeat-times: 5)]. Blast2GO Functional Annotation and Genomics Tool (https://www.blast2go.com/) was used for sequence annotation. Accordingly, the Identical Protein Groups Database of carrot was converted to a local peptide database using Blast2GO software. Integrated BLASTX algorithm was run with an e-value threshold of 1.00E-04 in order to translate the genomic sequences, perform alignments to the protein database, identify statistically significant matches and assign sequence descriptions based on protein annotations.
A similarity search was performed against a database of published carrot SSR markers [Citation2]. Toward this aim, carrot genomic sequences that contain the newly designed SSR markers were retrieved to Blast2GO software in FASTA format and converted to a local database. The sequences of 300 published carrot SSR loci [Citation2] were formatted as a single FASTA file and used as the query sequence to perform a BLAST search with the Megablast option.
DNA extraction, simple sequence repeat amplification and genotyping
Leaf tissue of 17 Turkish carrot landraces was obtained from Selçuk University, Department of Horticulture (Konya, Turkey). The carrot landraces were collected from Central Anatolia region (Konya and Ankara), the primary region of carrot cultivation in Turkey. DNA was extracted following a CTAB (cetyltrimethylammonium bromide) protocol modified from Doyle and Doyle [Citation37]. Accordingly, 100 mg batches of liquid nitrogen frozen, ground leaf tissue were used as starting material. The samples were incubated at 65 °C for 1 h in 800 μL of CTAB extraction buffer [100 mmol/L Tris-HCl (pH 8.0), 20 mmol/L EDTA (ethylenediaminetetraacetic acid, pH 8.0), 1.4 mol/L NaCl, 2% (w/v) CTAB, 1% PVP (polyvinylpyrrolidone)]. Following cell lysis, 600 μL of chloroform:isoamyl alcohol (24:1) was added prior to centrifugation at 14,000 rpm (Centrifuge 5810, Eppendorf, Hamburg, Germany) for 10 min at room temperature. The supernatant phase was taken, incubated with 200 μL of isopropanol at 4 °C for 60 min and DNA pellets were obtained as a result of centrifugation at 4 °C for 10 min at 14,000 rpm. A washing step was applied to the pellets using 100 μL of 70% ethanol for each sample, and DNA pellets were re-suspended in 100 μL of Tris-EDTA buffer (pH 8.0).
DNA samples were used to amplify 50 SSR markers randomly selected from the set of single-locus markers. PCR mixtures (25 μL) consisted of 1X Q5 Reaction Buffer (New England Biolabs Inc., Ipswich, MA), 0.2 mmol/L of each deoxyribonucleotide triphosphate (dNTP) (Promega Corp., Madison, WI), 0.5 U Q5 High-Fidelity DNA Polymerase (New England Biolabs Inc.), 0.50 μmol/L of each primer, and 5 ng template DNA. Standard thermal cycling conditions were used for all markers and consisted of one cycle of initial denaturation for 30 s at 98 °C, followed by 35 cycles of 98 °C for 10 s, 60 °C for 20 s, 72 °C for 30 s, with a final extension step of 2 min at 72 °C (T100 Thermal Cycler, Bio-Rad Laboratories Inc., Hercules, CA).
Marker fragments were run on a Qiaxcel Advanced capillary electrophoresis system (Qiagen, Hilden, Germany) following PCR amplification. A Qiaxcel DNA High Resolution Kit (Qiagen) capillary cartridge was used for capillary electrophoresis analyses. QX DNA Size Marker 25–500 bp, v2.0 (Qiagen) and QX Alignment Marker 15 bp/600 bp (Qiagen) were used for fragment sizing and size standard alignment, respectively. The OM800 high resolution method was utilized with a sample injection time of 10 s for capillary electrophoresis runs. Marker fragments were visualized and sized using QIAxcel ScreenGel Software (Qiagen). PIC (polymorphism information content) and He (expected heterozygosity) values were calculated for each polymorphic marker according to Botstein et al. [Citation38] and Nei [Citation39], respectively.
Results and discussion
Simple sequence repeat identification, primer design and analysis of cross-species transferability
In the present study, carrot genome assembly comprising 361.97 megabases (Mb) of sequence distributed over nine haploid chromosomes was mined for SSRs. A total of 138,759 repeat loci were identified in the carrot genome (). Dinucleotide repeats were the most abundant SSR type (75,633 SSRs), constituting 54.5% of the total number of identified SSRs (). A total of 54,988 mononucleotide repeat loci were identified, which represented the second most abundant SSR type (39.6%). The least common simple sequence repeat type in the dataset was hexanucleotide (). When the repeat loci identified in the carrot genome were classified based on motif frequency, the repeats of A/T nucleotides were the most abundant, representing 37.4% of all identified SSRs. Tandem repeats of AT and TA motifs together represented 32% of the entire pool of SSRs and their frequency in the set of repeat loci used for primer design reached almost 53% (). In addition, the three most abundant trinucleotide motifs were also AT-rich (). Thus, the results were consistent with other findings where genomes of dicotyledonous plants were analyzed for SSR abundance [Citation16,Citation21,Citation40–42].
Table 1. Simple sequence repeat (SSR) types in the carrot genome.
Table 2. Most abundant simple sequence repeat (SSR) motifs.
Despite the fact that mononucleotide SSRs represent the most abundant SSR type in several plant genomes, including tomato (Solanum lycopersicum), potato (Solanum tuberosum), pepper (Capsicum annuum), cucumber (Cucumis sativus), Arabidopsis (Arabidopsis thaliana) [Citation42], sesame (Sesamum indicum) [Citation41], purple false brome (Brachypodium distachyon), sorghum (Sorghum bicolor), rice (Oryza sativa), barrel clover (Medicago truncatula) and poplar (Populus trichocarpa) [Citation40], they are not the ideal targets to be converted to PCR markers. Genotyping mononucleotide SSRs can be error prone and mononucleotide repeats are more likely to represent noncoding regions since they are selected against in coding sequences [Citation43]. Therefore, mononucleotide SSRs were excluded from the marker design process and SSRs of two to six nucleotides (83,771 SSRs) were used for designing PCR primers (). Frequencies of SSR types used for the marker design process () and their distribution over nine carrot chromosomes are provided in .
Table 3. Statistics on simple sequence repeat (SSR) marker numbers and locations in the carrot genome assembly.
The high-throughput marker design process produced a total of 55,386 primer pairs that are predicted to amplify 67,279 SSR loci, corresponding to an overall average density of 5.38 kilobases (kb)/marker interval (). Marker loci were evenly distributed across the nine carrot chromosomes with marker densities ranging from 5.22 kb/marker interval (chromosome 6) to 5.47 kb/marker interval (chromosome 3) (). The start and end positions of the SSR loci in the chromosome sequences, the repeat motifs, number of repeats, flanking primer sequences and annealing temperatures can be accessed at https://figshare.com/articles/Marker_data/8593337/2 as ‘SSR Locus Information and Primer Data.xls’.
Genomic SSR markers developed in the present study were compared to the set of 300 markers formerly introduced by Cavagnaro et al. [Citation2]. As a result of the BLAST analysis, 163 statistically significant matches were obtained that displayed at least 90% similarity with the set of existing markers. Information for the set of common markers is available at https://figshare.com/articles/Marker_data/8593337/2 as ‘Common Marker Information.xls’.
The e-Mapping algorithm was run in order to predict the amplification profiles of the newly designed markers. The process revealed 51,160 single-locus markers plus 4226 primers that anneal at multiple genomic loci. The 51,160 loci still represented the carrot genome in high resolution with an average distance of 7.08 kb between adjacent markers (). Cavagnaro et al. [Citation2] reported a similar SSR frequency of 134.5 SSR/Mb (7.44 kb/marker interval) as a result of mining for SSRs (excluding mononucleotide repeats) in a database of carrot BAC-end sequences. The detailed results of the e-Mapping process including start and end positions of amplicons on nine reference assembly chromosomes and expected allele sizes for the single-locus SSR markers can be accessed at https://figshare.com/articles/Marker_data/8593337/2 as ‘e-Mapping Data.xls’.
In order to perform a genome-scale analysis of cross-species marker transferability, the GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) was searched for entries of assembly level Apiaceae genome sequences. Apart from carrot, a genome assembly record existed only for fennel, which included 968.14 Mb of sequence data organized as 300,377 assembled scaffolds. Fennel is an important cultivated member of the Apiaceae subfamily Apioideae together with carrot and other widely known vegetables and spices including celery, parsley, cumin, anise, coriander, parsnip and dill. In case an abundance of common markers are available for Apiaceae, they can be effectively utilized for genome evolution and synteny studies and, comparative mapping and cloning of identified genes. Therefore, it was valuable to assess the transferability of the newly developed carrot genomic SSRs to fennel. High-stringency cross-species mapping parameters were applied in order to ensure high confidence in detecting transferable markers. As a result, 794 carrot SSR markers were mapped on the fennel genome. The marker set was filtered for low-copy loci, resulting in a total of 578 markers that produced ≤ 3 alleles (). The list of the marker loci can be accessed at https://figshare.com/articles/Marker_data/8593337/2 as ‘Transferable Markers.xls’.
Table 4. Statistics of cross-species marker transferability to fennel genome assembly.
Annotation of simple sequence repeat loci
The newly designed SSR markers were annotated based on matching protein identities. Toward that direction, the Identical Protein Groups Database of carrot, which includes 58,303 protein records, was utilized to create a local database for annotating SSR-containing sequences. Carrot genomic sequences that allowed primer design were subjected to a high-throughput annotation analysis, resulting in 20,967 statistically significant matches in the database of protein sequences. Thus, 30.4% of the marker loci corresponded to predicted protein-coding genomic sequences. Among the matching carrot proteins, 13,920 entries (66.4% of the total number of matching peptides) had functional annotations, whereas the rest of the matching peptide sequences corresponded to uncharacterized or hypothetical functions. The matching protein descriptions of 20,967 carrot genomic SSR loci can be accessed at https://figshare.com/articles/Marker_data/8593337/2 as ‘Annotated Simple Sequence Repeat Loci.xls’.
Experimental validation of marker design and mapping processes
Out of the 51,160 single-locus SSR markers, 50 markers were experimentally evaluated. As a result, while all 50 primer pairs produced fragments within the expected size range (± 50 bp from the e-Mapping results), 46 markers (92%) produced amplicons from all 17 carrot accessions (). The amplification profiles of only five markers indicated multiple primer annealing sites, corresponding to an accuracy rate of 90% in predicting the amplification profiles by e-Mapping. In similar works, the amplification success of SSR markers designed through high-throughput, in silico sequence mining approaches was evaluated and reported by several research groups. For example Han et al. [Citation17] identified 364,347 SSRs in wheat genomic sequences, designed 295,267 SSR markers and experimentally validated 45 randomly selected markers on 23 wheat accessions. As a result of their work, the authors reported an amplification success rate of 71.1%. Liu et al. [Citation14] identified 101,157 SSR loci in a cucumber genome sequence and experimentally validated 50 SSR markers on 39 cucumber samples. The authors reported an amplification success rate of 100%. In another work, Dossa et al. [Citation22] identified 138,194 SSR loci in a sesame genome assembly, utilized the e-PCR algorithm for in silico marker amplification and experimentally validated 23 SSR markers on 48 sesame accessions. As a result, 91.3% of the markers successfully produced amplicons. Cui et al. [Citation21] designed 138,727 SSR markers using the bitter melon genome sequence and verified the amplification efficiency of 71 primer pairs on two accessions. Wang et al. [Citation23] designed 1,224,048 primer pairs that amplify SSR loci from seven Nicotiana genomes and experimentally evaluated 120 SSR markers on five Nicotiana accessions. All tested primers produced amplicons from at least four out of the five accessions. Similar results were obtained in the course of the present study (amplification success and accuracy in predicting amplification profiles both exceeding 90%) and it was feasible to verify the efficiency of the high-throughput marker design and in silico mapping analyses. Moreover, 28 out of 46 markers (61%) yielded polymorphisms among the 17 carrot landraces. Amplification information for the experimentally evaluated markers can be accessed at https://figshare.com/articles/Marker_data/8593337/2 as ‘Evaluated Markers.xls’. Twenty-five of the polymorphic markers were codominant, whereas three were dominant (Chr3-MK592, Chr3-MK745, Chr9-MK959). The average number of alleles observed for the codominant SSR loci was 3.1. The minimum and maximum He values of the codominant markers were 0.46 and 0.78 respectively, and the average He was 0.58 (). The average PIC value of the 25 codominant markers was 0.49 and ranged from 0.35 to 0.75 (). Despite the limited geographical distribution of the carrot landraces used in the study, a significant rate of polymorphism (61%) was observed for the markers and the average PIC value, the mathematical measure of marker informativeness, was moderate. Thus, it was valuable to observe that the SSR loci identified in the present work had potential to reveal genotypic diversity in carrot.
Figure 1. A set of polymorphic marker fragments visualized by high-resolution capillary electrophoresis. Electropherograms of different size alleles amplified from carrot landraces are displayed for each marker. Primer sequences of markers can be accessed at https://figshare.com/articles/Marker_data/8593337/2.
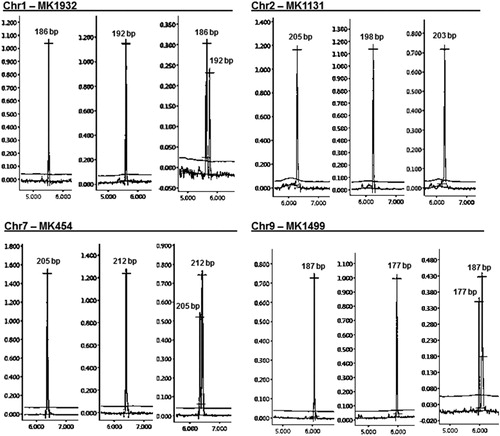
Table 5. Informativeness of the codominant, polymorphic markers.
Conclusions
Marker-assisted breeding has not been as widely utilized for carrot and other Apiaceae species as it is for other agriculturally relevant families, namely Solanaceae, Fabaceae, Brassicaceae and Poaceae. This is due to the fact that molecular genetic resources including DNA markers are not as abundant for Apiaceae. In this study, the carrot physical chromosomes were saturated with sequence-based markers that actually constitute a dense ‘physical map’ of the carrot genome. In addition, the potential cross-species transferability of the markers within Apiaceae members was demonstrated with the identification of a large set of markers mapped on the fennel draft genome sequence. Moreover, annotation analysis provided information on 13,920 markers that reside in protein-coding sequences, thus, displayed the potential of the markers for gene tagging and functional genomics. The present work is the first time that a large collection of carrot-specific SSR markers with known positions on carrot physical chromosomes is made available for carrot molecular genetics. It is expected that the markers introduced in this study will be practical genomic tools useful for gene/quantitative trait loci mapping, functional genomics, germplasm characterization and preservation, and molecular breeding studies in carrot.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
All data generated during this study are available at Figshare (https://doi.org/10.6084/m9.figshare.8593337).
Related Research Data
References
- Iorizzo M, Senalik DA, Grzebelus D, et al. De novo assembly and characterization of the carrot transcriptome reveals novel genes, new markers, and genetic diversity. BMC Genomics.2011;12(1):389.
- Cavagnaro PF, Chung SM, Manin S, et al. Microsatellite isolation and marker development in carrot—genomic distribution, linkage mapping, genetic diversity analysis and marker transferability across Apiaceae. BMC Genomics.2011;12(1):386.
- Cavagnaro PF, Chung SM, Szklarczyk M, et al. Characterization of a deep-coverage carrot (Daucus carota L.) BAC library and initial analysis of BAC-end sequences. Mol Genet Genomics. 2009;281(3):273–288.
- Iorizzo M, Ellison S, Senalik D, et al. A high quality carrot genome assembly provides new insights into carotenoid accumulation and asterid genome evolution. Nat Genet. 2016;48(6):657–666.
- Baranski R, Allender C, Klimek-Chodacka M. Towards better tasting and more nutritious carrots: Carotenoid and sugar content variations in carrot genetic resources. Food Res Int.2012;47(2):182–187.
- Akhtar S, Rauf A, Imran M, et al. Black carrot (Daucus carota L.), dietary and health promoting perspectives of its polyphenols: A review. Trends Food Sci Technol. 2017;66:36–47.
- Simon PW. Domestication, historical development, and modern breeding of carrot. In: Janick J, editor. Plant breeding reviews. Vol. 19. New York: Wiley; 2000. p. 157–185.
- Wang F, Wang GL, Hou XL, et al. The genome sequence of ‘Kurodagosun’, a major carrot variety in Japan and China, reveals insights into biological research and carrot breeding. Mol Genet Genomics. 2018;293(4):861–871.
- Morgante M, Hanafey M, Powell W. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat Genet. 2002;30(2):194–200.
- Victoria FC, Da Maia LC, De Oliveira AC. In silico comparative analysis of SSR markers in plants. BMC Plant Biol. 2011;11(1):15.
- Boccacci P, Beltramo C, Sandoval Prando MA, et al. In silico mining, characterization and cross-species transferability of EST-SSR markers for European hazelnut (Corylus avellana L.). Mol Breed 2015;35:21.
- Zhang XF, Sun HH, Xu Y, et al. Development of a large number of SSR and InDel markers and construction of a high-density genetic map on a RIL population of pepper (Capsicum annuum L.). Mol Breed 2016;36:92.
- Agarwal G, Sabbavarapu MM, Singh VK, et al. Identification of a non-redundant set of 202 in silico SSR markers and applicability of a select set in chickpea (Cicer arietinum L.). Euphytica 2015;205(2):381–394.
- Liu J, Qu J, Hu K, et al. Development of genomewide simple sequence repeat fingerprints and highly polymorphic markers in cucumbers based on next-generation sequence data. Plant Breed. 2015;134(5):605–611.
- Bhattarai G, Mehlenbacher SA. In silico development and characterization of tri-nucleotide simple sequence repeat markers in hazelnut (Corylus avellana L.). PLoS ONE. 2017;12(5):e0178061.
- Cavagnaro PF, Senalik DA, Yang L, et al. Genome-wide characterization of simple sequence repeats in cucumber (Cucumis sativus L.). BMC Genomics.2010;11(1):569.
- Han B, Wang C, Tang Z, et al. Genome-wide analysis of microsatellite markers based on sequenced database in Chinese spring wheat (Triticum aestivum L.). PLoS ONE. 2015;10(11):e0141540.
- Parida SK, Verma M, Yadav SK, et al. Development of genome-wide informative simple sequence repeat markers for large-scale genotyping applications in chickpea and development of web resource. Front Plant Sci. 2015;6:645
- Dettori MT, Micali S, Giovinazzi J, et al. Mining microsatellites in the peach genome: development of new long-core SSR markers for genetic analyses in five Prunus species . Springerplus. 2015;4:337
- Zhu H, Guo L, Song P, et al. Development of genome-wide SSR markers in melon with their cross-species transferability analysis and utilization in genetic diversity study. Mol Breed 2016;36:153.
- Cui J, Cheng J, Nong D, et al. Genome-wide analysis of simple sequence repeats in bitter gourd (Momordica charantia). Front Plant Sci. 2017;8:1103
- Dossa K, Yu J, Liao B, et al. Development of highly informative genome-wide single sequence repeat markers for breeding applications in sesame and construction of a web resource: SisatBase. Front Plant Sci. 2017;8:1470
- Wang X, Yang S, Chen Y, et al. Comparative genome-wide characterization leading to simple sequence repeat marker development for Nicotiana. BMC Genomics.2018;19(1):500.
- Xia X, Luan LL, Qin G, et al. Genome-wide analysis of SSR and ILP markers in trees: diversity profiling, alternate distribution, and applications in duplication. Sci Rep. 2017;7(1):17902.
- Baranski R, Maksylewicz-Kaul A, Nothnagel T, et al. Genetic diversity of carrot (Daucus carota L.) cultivars revealed by analysis of SSR loci. Genet Resour Crop Evol. 2012;59(2):163–170.
- Ipek A, Turkmen O, Fidan S, et al. Genetic variation within the purple carrot population grown in Ereğli district in Turkey. Turk J Agric For 2016;40:570–576.
- Ma ZG, Kong XP, Liu LJ, et al. The unique origin of orange carrot cultivars in China. Euphytica 2016;212(1):37–49.
- Mandel J, Ramsey AJ, Iorizzo M, et al. Patterns of gene flow between crop and wild carrot, Daucus carota (Apiaceae) in the United States. PLoS ONE. 2016;11(9):e0161971.
- Yildiz M, Willis DK, Cavagnaro PF, et al. Expression and mapping of anthocyanin biosynthesis genes in carrot. Theor Appl Genet. 2013;126(7):1689–1702.
- Budahn H, Barański R, Grzebelus D, et al. Mapping genes governing flower architecture and pollen development in a double mutant population of carrot. Front Plant Sci. 2014;5:504
- Parsons J, Matthews W, Iorizzo M, et al. Meloidogyne incognita nematode resistance QTL in carrot. Mol Breed 2015;35:114.
- Iorizzo M, Senalik DA, Ellison SL, et al. Genetic Structure and domestication of carrot (Daucus carota subsp. sativus) (Apiaceae). Am J Bot.2013;100(5):930–938.
- Stelmach K, Macko Podgorni A, Machaj G, et al. Miniature inverted repeat transposable element insertions provide a source of intron length polymorphism markers in the carrot (Daucus carota L.). Front Plant Sci. 2017;8:725
- Grzebelus D, Iorizzo M, Senalik D, et al. Diversity, genetic mapping, and signatures of domestication in the carrot (Daucus carota L.) genome, as revealed by Diversity Arrays Technology (DArT) markers. Mol Breeding. 2014;33(3):625–637.
- Wang X, Wang L. GMATA: An integrated software package for genome-scale SSR mining, marker development and viewing. Front Plant Sci. 2016;7:1350
- Schuler GD. Sequence mapping by electronic PCR. Genome Res. 1997;7(5):541–550.
- Doyle JJ, Doyle JL. Isolation of plant DNA from fresh tissue. Focus 1990;12:13–15.
- Botstein D, White RL, Skolnick M, et al. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 1980;32(3):314–331.
- Nei M. Analysis of gene diversity in subdivided populations. PNAS 1973;70(12):3321–3323.
- Sonah H, Deshmukh RK, Sharma A, et al. Genomewide distribution and organization of microsatellites in plants: An insight into marker development in Brachypodium. PLoS ONE.2011;6(6):e21298.
- Uncu AO, Gultekin V, Allmer J, et al. Genomic simple sequence repeat markers reveal patterns of genetic relatedness and diversity in sesame. The Plant Genome. 2015;8(2):0.
- Cheng J, Zhao Z, Li B, et al. A comprehensive characterization of simple sequence repeats in pepper genomes provides valuable resources for marker development in Capsicum. Sci Rep. 2016;6(1):18919.
- Gu T, Tan S, Gou X, et al. Avoidance of long mononucleotide repeats in codon pair usage. Genetics 2010;186(3):1077–1084.