
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Poly ADP-ribose polymerases (PARPs) are family of proteins that use nicotinamide adenine dinucleotide (NAD) as substrate. Seventeen putative PARP sequences were determined in the human genome. Although PARPs show a variety of functions and low sequence identities, they share common structural and functional properties. In our study, PARP1 and PARP2 and PARP4 sequences in different species were compared; it was found that active sites of PARP1 for human, rat and mouse have highly conserved sequence. Overall folding of PARP1, PARP2 and PARP4 confirms similarity in catalytic domains but can differ in substrate proteins. The three-dimensional structure of PARP4 was interacted with NAD using the molecular docking method and the interaction sites were determined. When we modeled the three-dimensional structure of PARP4 using MODELLER v9.22 algorithm and examined the interaction with Autodock v4.2 in computer environment, we observed that the enzyme is connected with a common motif similar to PARP1 and PARP2. When PARP1 and PARP2 interact with this common motif with NAD, we experimentally observed that these structures interact directly with NAD in order to undergo catalytic reactions by Thermal-Shift assay. The PARP4–NAD complex with the binding energy −26.73 kJ/mol was further used for molecular dynamics analysis. Root mean square deviation (RMSD) for all backbone atoms, electrostatic energy, van der Waals energy of PARP4-NAD complex were studied in the form of molecular dynamics trajectories to throw light on the medically important PARP family of enzymes.
Introduction
ADP-ribosylation is a highly dynamic post-translational modification mechanism. Nicotinamide adenine dinucleotide (NAD) is an important coenzyme involved in redox reactions and the transfer of the reduction potential between molecules in the intracellular electron transport system. A group of NAD-metabolizing enzymes are poly (ADP-ribose) polymerases (PARPs). PARPs catalyze the breakup of NAD into nicotinamide and ADP-ribose by transferring ADP-ribose to a target protein subsequently [Citation1]. This ability of the PARP family of enzymes is based on catalyzing the transfer of ADP-ribose to target proteins. PARPs take part in various important cellular processes such as modulation of chromatin structure, transcription, replication, recombination [Citation2]. PARPs are involved in cellular processes of most eukaryotic cells and function in posttranslational protein modifications known as an instance in which the ADP-ribose moiety is transferred from NAD onto specific substrates. PARP has seventeen isoforms that have structural domains and functions have been identified [Citation3, Citation4]. Studies have shown that PARP-1 is a key player in DNA damage repair, maintenance of genomic integrity and mammalian longevity and the most plentiful isoform that is involved in chromatin structure and DNA metabolism [Citation5, Citation6]. PARPs modify numerous cellular processes such as DNA repair, energy metabolism, cell proliferation and differentiation, apoptosis or immune functions [Citation1, Citation7]. Activation of PARP causes decomposition of NAD into nicotinamide and ADP-ribose; this system takes place through the consumption of NAD. Through the excessive activation of PARP, the number of ATP molecules decreases together with reduction in the amount of ATP molecules and these processes result in inflammatory injury, cell dysfunction, and ultimately necrotic cell death [Citation5, Citation8]. PARPs are important targets for anticancer therapies because they prevent repairing DNA damage in cells and cause apoptosis [Citation9, Citation10]. Different phase I and phase II clinical trials are currently continuing for the evaluation of the usage of PARP inhibitors for therapy of ovarian cancer, breast cancer, prostate cancer, metastatic melanoma, collateral cancer, gastric cancer, hepatocellular carcinomas, sarcomas, lymphomas, pancreatic neoplasms and glioblastomas [Citation11, Citation12]. PARPs are up-and-coming molecules for the development of inflammation, degenerative, vascular diseases, and ischemia and cancer drugs [Citation13]. Vault poly (ADP-ribose) polymerase (VPARP; PARP4) is identified as the largest member of the PARP family at 193 kDa [Citation14]. PARP4 is a cytoplasmic ribonucleoprotein complex that executes PARP activity and PARylates major vault protein (MVP) more than itself [Citation15]. PARP4 is also catalytically active like PARP1 and PARP2 mostly in humans, associated with vaults, which are very large cytoplasmic ribonucleoprotein particles, and exist in a non-vault associated form in eukaryotic cells [Citation16]. Vaults are studied best in mammals and they are composed of three proteins: major vault protein (MVP, 100 kDa), telomerase-associated protein (TEL1, 240 kDa) and vault poly(ADP-ribose) polymerase (VPARP, 193 kDa, 1724 amino acids). There are several protein–protein interaction domains of VPARP that involve interactions with other vault components and stabilize the structure of vaults [Citation14, Citation17]. It is known that PARP4 is an active enzyme and the function of such localized PARP activity is still elusive. Zon et al. [Citation18] reported a complex relationship, direct and dynamic, between vaults and PARP4.
The role of PARP4 in intracellular transport can be analyzed more detail by taking these studies as reference. In our interaction analyses, we predicted the three-dimensional (3D) structure of PARP4 and we compared the obtained structure to PARP1 and PARP2. The common motif that identifies NAD and has 12 amino acids was also defined as a result of these comparisons. Using a molecular docking system, we demonstrate within PARP4 an intermolecular binding site and show that VPARP contains amino acid stretches mediating intramolecular binding. Molecular modeling suggests that PARPs use substrate assisted catalysis in order to activate the NAD substrate so PARP4’s signature of NAD fold is also shown.
Materials and methods
Homology modeling and model evaluation
The 3D structure of PARP4 was built using homology modeling and the information in Protein Data Bank. NCBI (National Center for Biotechnology Information, https://www.ncbi.nlm.nih.gov/pubmed/) protein sequence database and template was defined using PSI-BLAST (Position-Specific Iterative Basic Local Alignment Search Tool, https://blast.ncbi.nlm.nih.gov/) against the RCSB (Research Collaboratory for Structural Bioinformatics, https://www.rcsb.org/) protein data bank. The sequence of PARP4 was revoked from this protein sequence database. The 3D-structure was constructed using MODELLER 9.18 (http://salilab.org/modeller/) algorithm. The structure was subjected to substantiation and evaluation by using the protein structure, structure evaluation and pattern estimation tools in the algorithm. Valuation graphical plots were done by using Atomic Non-Local Environment Assessment (Anolea). Assessment of local and global regions in the model was done by using Qualitative Model Energy Analysis (Qmean) scoring. Gromos Molecular Dynamics simulations were used to analyze the experimental results. Unwanted interactions were removed by using GROMOS96 force field in MODELLER algorithm with energy minimization. RAMPAGE Ramachandran Plot Analysis online tools (http://mordred.bioc.cam.ac.uk/∼rapper/rampage.php) was used to draw Ramachandran Plots, visualize energetically favoured regions and analyze folding angles of PARP4. PROCHECK was used to analyze the stereochemistry of PARP 4 and to assess the quality of the structure.
Molecular dynamics (MD) simulations
We obtained the best model by applying a docking procedure with the MD simulation to refine the protein interface structure. Nevertheless, during the simulation, explicit constraint functions were not used to maintain the initial docking contacts [Citation19]. Initially the energy was minimized using 1000 steps of steepest descents and conjugate gradient was minimized in 2000 steps for structures by using Kollman all-atom force field. Dielectric constant and unbounded cut off were set to 1 and 8 A respectively and dielectric function was distance dependent. In order to remove unrealistically close steric clashes and large deviations that arise from ideal geometry of conformational changes of amino acid side chains after docking, energy minimization with classical force field was applied [Citation20]. The starting structure of the MD simulation was the energy minimized structure. NAMD (Not (just) Another Molecular Dynamics program) molecular simulation package was used for all MD simulations. NAMD version 2.9 was used for analyzing the interactions between PARP4 and NAD. PARP4 molecular dynamics (MD) simulations had 300 K mean temperature and pH 7.0. In order to obtain high-performance simulation of large biomolecular systems, a parallel molecular dynamics code is designed called NAMD [Citation21]. To identify specific interactions at the interface, compute inter-residue distances and other calculations, MD simulation was used and the final structure built.
Molecular docking
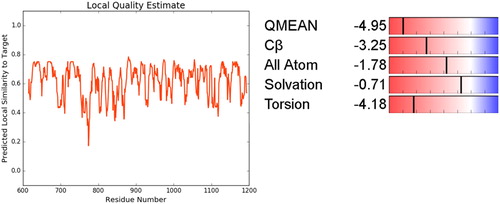
The acquired data of modeled structures (pdb-format) from DataBank with the supplementary data for PARP4 were obtained from these data by using Autodock. Lamarck’s mechanism was used for genetics modeling, which is the idea of the inheritance of acquired characters. Kollman united atom charges, solvation parameters and polar hydrogen were added to the protein and Gastegier charges were used in Autodock. Distance between grid points was set at the 0.375 Å default value. The x, y and z axes of grid boxes were adjusted as 480 Å, 260 Å, 280 Å respectively. These dimensions contained all of the amino acid residues of PARP4 and the sizes of grids were large enough to include all the domains of PARP4. Our first purpose was to decode the binding domain The step size was 0.2 Å and 50 runs of simulation were done. Torsion and orientation angle were 50. The other adjusted data values were 1000.0 for the grid energy, 0.5 Å for the cluster tolerance, 0.8 for the crossover rate, 0.02 for mutation rate, 1 for maximum number of top individuals and 1000 for the maximum number of generations. QMEAN (Quantitative Model Energy Analysis) [Citation22], which is a linear combination of four potential statistical terms, was used to derive both global and local absolute quality estimates by one single model. QMEAN score was used for the results. Root mean square fluctuation (RMSF) analysis was performed to determine the flexibility of the molecules and to compare structural similarities. The structure similarity comparison was completed with root mean square deviation (RMSD) analysis.
Obtaining PARP1 and PARP2 subunits by trypsin partial digestion
PARP1 and PARP2 groups were partially digested in the presence of 50 mmol/L Tris-HCl with trypsin (molar ratio 1/200), 250 mmol/L sucrose, 7 mmol/L methionine (MET), 0.2 mmol/L phenylmethylsulfonyl fluoride (PMSF) at pH 7.4 for 60 min at 37° C. In order to stop the reaction, soybean meal-derived trypsin inhibitor was added at 1:1 ratio then denatured with sodium dodecyl sulfate (SDS). Trypsin-separated fractions were purified by polyacrylamide gel electrophoresis (SDS-PAGE) and chromatographic methods. The samples were concentrated in protein concentration spin columns (Vivaspin-10.000). The amount of ADP-ribosylation activity of the obtained samples was determined by enzyme-linked immunosorbent assay (ELISA). A 96-well colorimetric assay kit was used to measure the activity of PARPs (HT universal colorimetric PARPs assay kit) with histone-coated strip wells (Trevigen, USA) according to the manufacturer’s instructions.
Chromatography analysis
HiPrep Sephacryl S-100 (16 × 60 cm) column was used for filtration chromatography in the study. The total volume (Vc) of the column was 120 mL and the dead volume (V0) was 40 mL. The column was calibrated with aprotinin (6.5 kDa), carbonic anhydrase (29 kDa), ovalbumin (43 kDa), BSA (66 kDa) and conalbumin (75 kDa) standards and gel-phase distribution coefficients (µ) were calculated according to arrival volume to obtain a calibration graph
The protein samples to be studied were separated into fragments and the arrival volumes of these fragments were checked from the calibration graph. Optical densities of the samples were determined at 280 nm, 0.35 mPa pressure and 0.8 mL/min velocity and after separation as 0.5 mL.
Electrophoresis analysis
Samples were tested by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) according to the Laemmli method. A 12% separation gel was used. The samples were mixed with a denaturing buffer in a 1: 1 ratio and were boiled for 2 min. The electrophoresis tank was filled with run-off buffer. A multi-protein standard (Fermentas Protein Ladder) with molecular weight between 10 kDa and 260 kDa was used as a molecular weight standard. The gel was stained with coomassie brillant blue. Then excess dye was removed from the gel in the presence of 7% acetic acid and 30% methanol. The gel, which had been stained with protein bands, was placed on a 3 mm Whatman filter paper, wrapped with stretch film, and dried by applying vacuum for 1 h at 80 °C in a gel drier.
Thermal shift assay
Thermal Shift Assay was used to screen the thermal denaturation of proteins in the presence of fluorescent dye (SYPRO orange) and using a typical real-time polymerase chain reaction (PCR) instrument. Fluorescent intensity was plotted as a function of temperature to obtain a sigmoidal curve defined by a two-state transition. The turning point (Tm) of the transition curve was calculated by Boltzmann equation. For the analysis, PCR device and the corresponding “96 well plate” were used. The experiment was repeated 3 times for each sample. Before addition of SYPRO Orange, PARPs was applied at 0.01, 0.025, 0.05, 0.1 and 0.5 µg/mL concentrations to the PARPs applied groups. The fluorescence intensity was measured at 450–490 nm excitation, 560–580 nm detection with a 15-second time period between 15 °C to 90 °C with an increment of 0.5 °C. Based on the thermal stability values measured in the linear zone, the Tm values from the Boltzman equation were calculated with MS Excel. After application of SYPRO Orange, 10 µg/mL PARP1 and NAD in increasing concentrations, fluorescence intensity was measured between 20 °C and 90 °C temperature values at 0.5 °C intervals with a period of 15 s (0.5 °C/10 s) at 580 nm wavelength. NAD was not applied to the control group. Tm values measured after the PARP2-NAD interaction were calculated with GraphPad and Excel from the thermal stability graphs. The denaturation midpoint temperature (Tm) values were calculated from the linear zone by using the Boltzmann equation.
Results and discussion
PARP1 and PARP2 data files of human and different species were obtained from available databases. PARPs are enzymes that are found in most eukaryotes and are known to function in post-translational protein modifications. PARPs share a specific catalytic domain with a characterized PARP motif [Citation16]. PARP1 sequences in different species were compared, considering that the common motif that recognizes NAD might be similar between species.
As a result of the comparisons on PARP1, some hot spots were predicted that have binding areas (active sites) and binding sites (hot spots) with binding energies higher than the others (). The same analysis procedure was repeated in PARP2. Active regions were identified for human and mouse amino acid sequences (). The results indicated that the predicted active sites of PARP1 in human, rat and mouse have highly conserved sequences.
Figure 1. Comparison of common structural indexes in PARP-1 in four different species. Note: H = Human, B = Bovine, M = Mouse, R = Rat.
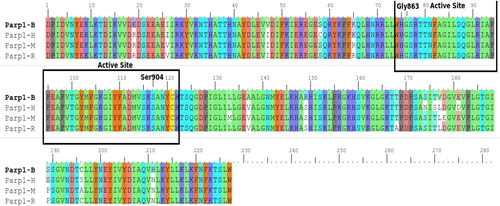
Figure 2. Comparison of PARP-2 amino acid sequences in human and mouse. Note: H = Human, M = Mouse.
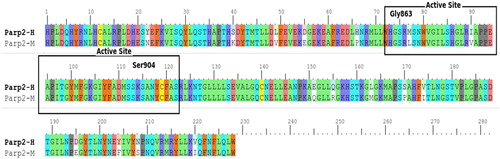
The predicted active sites of PARP2 differed in positions 195, 204, 208 and 220 for human and mouse. Homology modeling and docking studies of the protein were carried out to understand the role of PARP4 and its interactions.
The template search programs revealed several sequence homologues to PARP4 with experimental 3D protein structures within the Protein Data Bank (PDB 6FPY_A). The identity percentage and E-values (expectation values) for the best hit template protein were obtained. Pairwise alignment between PARP4 and 6FPY_A is shown in and the generated scores and conserved residues are given in .
Figure 3. Sequence alignment of PARP4 with template PDB ID 6fpy.

Figure 4. Local Similarity and Qmean analysis of PARP-4.
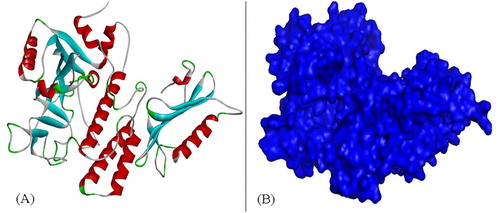
After active site comparisons of PARP1 and PARP2, we modeled PARP4, which is structurally similar to these PARPs, but also includes some differences. In PARP4, the structure and active regions were modeled using the homology modeling method (). The results of the blast scan showed that mold PDB files had at least 52% similarity. The overall folding of PARP1, PARP2 and PARP4 confirms similarity in catalytic domains but can differ in substrate proteins. Similar and different domains are expected to occur because even plant and human PARP proteins have similar and different regions when compared [Citation23]. However, the discovery of these regions can shed light on some experimental results.
Figure 5. Secondary structure (A) and surface structure (B) of PARP4.
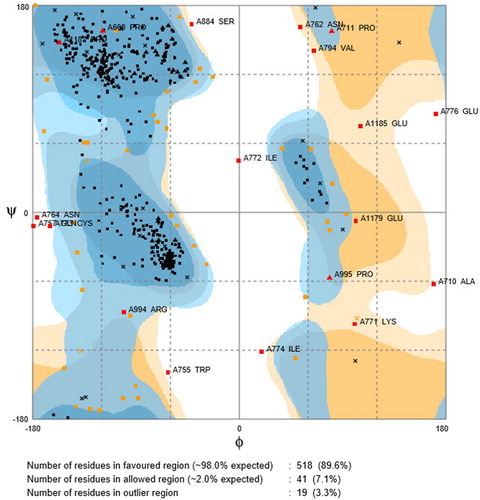
After the modeling, the Ramachadran plot showing the folding map in the model was drawn. PROCHECK depicted 89.6% in the core region () and was the optimal score. The Ramachandran plot of PARP4 showed torsion angles and as a result of collision between atoms, disallowed rotations of excluded regions. Blue shades on Chi-Chi2 state favorable regions enclosed by residues on plot and yellow shades show residues in outlier region.
Figure 6. Ramachandran plot for PARP4, Ramachandran contour plot depicting Φ and Ψ angles. Source: http://mordred.bioc.cam.ac.uk/∼rapper/rampage.php.
To identify the most possible conformation and folding analysis of PARP4, molecular dynamics simulation analysis was executed by using TR-Data Grid system (TUBITAK) (). PARP4 motif was subjected to flexibility (RMSF) analyses () and the stereochemistry provided structure was added. The three-dimensional structure of PARP4 was interacted with NAD using the molecular docking method and the interaction sites were determined ().
Figure 7. Molecular dynamics calculations of PARP4.
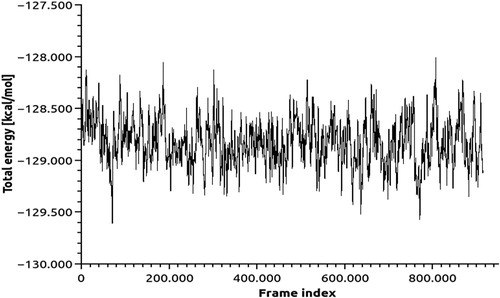
Figure 8. Flexibility calculations of PARP4 RMSF analysis.
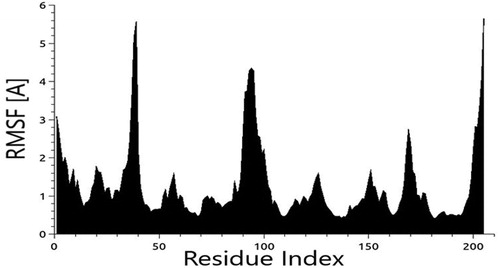
Figure 9. Interaction residue (A) and area (B) of PARP4-NAD.
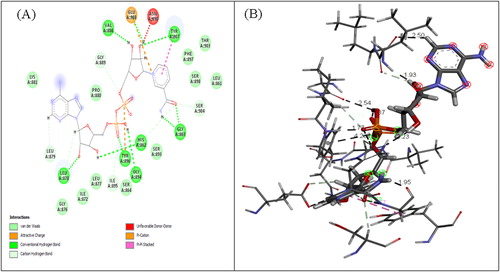
Docking study between PARP4 and NAD revealed significant contribution of hydrogen bonds, attractive van der Waals, repulsive van der Waals, atomic contact energies and global interaction energy of −7.32, −34.18, 17.12, 18.09 and −27.54 (kJ/mol) respectively. The PARP4–NAD complex with the binding energy −26.73 kJ/mol was further used for carrying out MD. RMSD for all backbone atoms, electrostatic energy, van der Waals energy of PARP4-NAD complex were studied in the form of MD trajectories.
The obtained PARPs data files were compared with each other to determine the common active site (). The identified common motif that recognizes and catalyzes NAD was determined (). The primary structure of PARP1 is compatible with the crystal structures of PARP1 domains of the model proposed by Langelier et al. [Citation24] and human PARP-2 in corresponding to the structural characteristics reported by Yelamos et al. [Citation16].
Figure 10. Active site determination based on common structural motif that recognizes the NAD region of PARPs belonging to human, rat and mouse.
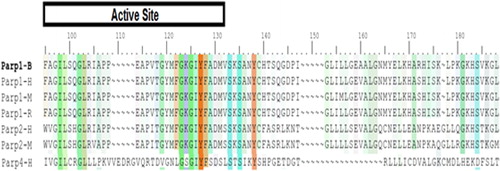
Figure 11. Folding analysis of PARPs (1, 2, 3, 4, 6, 11) binding NAD.
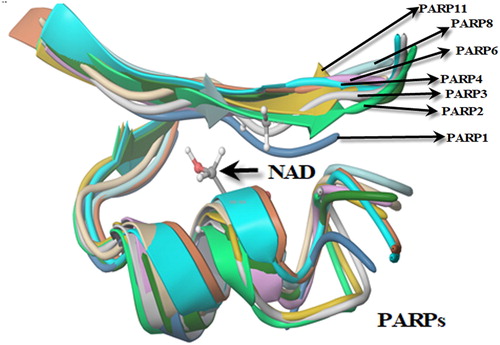
Figure 12. Secondary structure of PARP1 (A) and PARP2 (B) with NAD interaction.
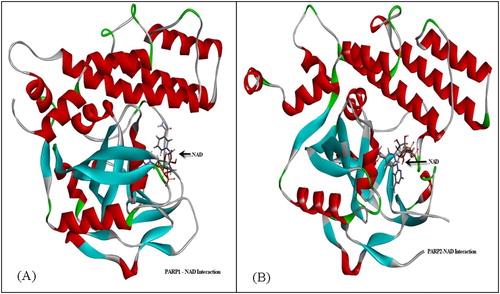
PARP1 and PARP2 were interacted with NAD (). The molecular weights of PARP1 and PARP2 were obtained with electrophoresis and chromatography analysis and were found to be similar to previous reports [Citation25–27]. Then they were interfered with NAD and the theoretically determined regions were tested. PARP1 and PARP2 are among the most studied proteins and they create poly(ADP-ribose). However, their ADP-ribose binding domains are not determined. PARP4 does not generate poly(ADP-ribose); it attaches ADP ribose as a monomer but its ADP-ribose binding domains also are not determined [Citation28]. In this study, we predicted the NAD binding domains of PARP1, PARP2 and PARP4, which is important for preclinical experimental procedures ( and ).
We prepared a series of data sets for the PARP1, PARP2 and PARP4 proteins before interacting with NAD in the simulation under optimal conditions. In this study, receptor and ligand (NAD) grids were formed for the data sets. Similarity (RMSD) calculations for all three PARP proteins were deduced ().
Table 1. Comparative RMSD values of PARP1, PARP and PARP4 (°A).
After obtaining data sets, docking was performed to determine the interaction regions of the common motif that can recognize the NAD molecule for the PARPs to catalyze the NAD reaction and to determine the binding energy necessary to connect this common motif to the NAD region of the interaction interface. The potential energies were calculated in order to determine the highly probable interaction regions by using scoring techniques as a result of the docking process. Alemasova and Lavrik’s review shows the NAD-binding site of PARP1 [Citation29]; however there is no simulation for NAD-bound PARP2 and PARP4.
During calculation of the inter- associations between structures, bond elasticity, bending angles, dihedral angles and interactions of amino acids with H atoms in the liquid medium in which they exist, Van der Waals interactions between atoms forming amino acids and electrostatic interactions resulting from the charge of atoms were considered. At the end of the study, the regions with the highest probability of interaction with NAD and the amino acid sequences belonging to these regions were determined from PARP groups ().
Table 2. Catalytic sites from PARPs with the highest probability of interaction with NAD.
Experimental studies were made in accordance with the theoretical information from simulations. Western Blot () and chromatography () analyses were performed for the purity of PARP1 and PARP2 enzymes to be studied experimentally.
Figure 13. Electrophoresis (A) and chromatography (B) analysis of PARP1 and PARP2. Note: For electrophoresis analysis, 5 μg protein was loaded to gel.
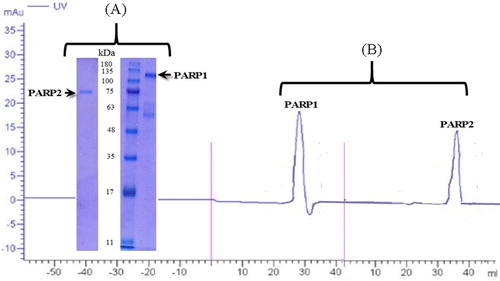
Electrophoresis and chromatographic analysis confirmed the purity of the commercially available PARP1 and PARP2 enzymes to be used in the following stages of the study. Chromatographic analyses of both molecules confirmed their molecular weights () [Citation25–27]. These enzymes were then interacted with NAD to proceed to the next step. Enzymes with known purity were tested for NAD interaction via thermal shift assay, which is a method used for protein–protein or protein–molecule interaction investigations. PARP1 and PARP2 have been shown to interact with NAD ( and ).
Figure 14. PARP1-NAD interaction was tested with thermal shift assay. Melting temperatures of PARP1-NAD interaction (Tm values).
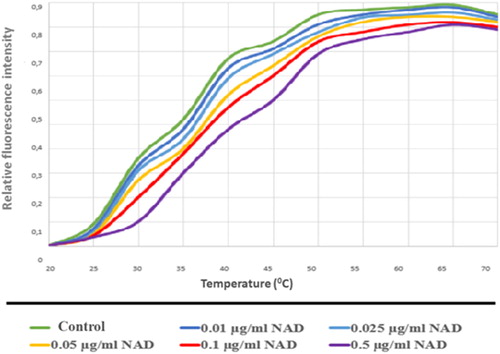
Figure 15. PARP2-NAD interaction was tested with thermal shift assay. Melting temperatures PARP2-NAD interaction (Tm values).
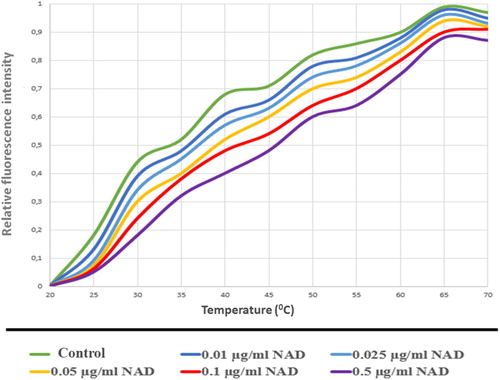
As a result of these assays, PARP1 and PARP2 proteins, which bind to NAD and carry out the catalysis reaction with the common structural motif, were determined to interact with NAD in increasing concentrations (). These results show usable values for NAD dependent PARylation.
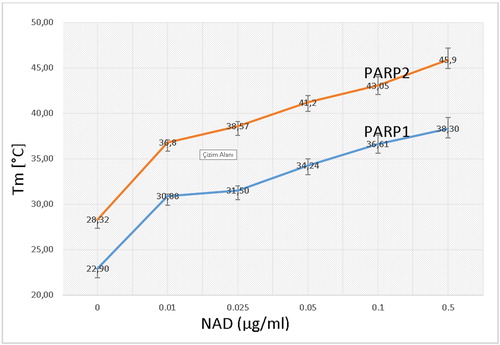
Figure 16. Interaction graph of PARP1 and PARP2 with NAD. (10 µg/ml PARP1, PARP2 and NAD in increasing concentrations).
PARP proteins catalyze the poly ADP-ribosylation of DNA-binding proteins to control their function in eukaryotes [Citation30]. The DNA strand breaks activate PARP enzyme which is responsible for DNA repair mechanisms. Mild genotoxic damage induces DNA repair by inducing PARP activation [Citation31]. In moderate cell damage, cells with DNA damage at irreversible levels are directed to the apoptosis [Citation32]. In the case of severe DNA damage, PARP is overactive and causes cell necrosis by killing intracellular energy metabolism [Citation33].
PARP enzymes share common structural and functional properties, although they display different sequence units and lower sequence identities. First, they catalyze a common reaction, ADP-ribosylation. Second, they have similar catalytic domains that bind NAD + in their tertiary structure.
We found that the poly (ADP-ribose) Polymerase 4 enzyme has a common structural NAD-binding motif with PARP1 and PARP2. We observed that these three enzymes with the same mechanism have a common 12-amino acid motif that binds to NAD, and that when NAD interacts with PARP1 and PARP2 at increasing concentrations, the amount of interacted NAD molecules also increases. These data reveal high activity of NAD and show specific catalysis by PARP1 and PARP2 for protein PARylation. PARP1, PARP2 and PARP4 structures are different in length and their NAD-binding sites vary in size and rigidity. The structural differences contribute to differential inhibitor binding [Citation28].
Inhibition of PARPs in cancer treatments is thought to regulate DNA repair and increase the effects of anticancer therapies [Citation34]. Structural similarities and differences among PARPs contribute the design of PARP inhibitors for potential clinical applications. Existing PARP inhibitors are designed to compete with NAD+ because of high sensitivity of NAD to PARP inhibitor treatment [Citation35]. Similar NAD binding parts in PARP1, PARP2 and PARP4, their catalytic domains, crystal structure and binding energy of PARP4 were shown as a contribution to PARP inhibitor developments.
Conclusions
Here, an important member of the PARP family, PARP4, was modeled with simulation modeling. As a result of the theoretical analyses, we shed light on the interaction of this structure with NAD by predicting the binding of this enzyme to NAD and detecting that it has a common structural motif with PARP1 and PARP2 enzymes. PARP4 binds to NAD with 12 amino acids (YFSDSLSTSIKY) in its three-dimensional structure. This was confirmed experimentally. Our data suggested that the crystal structure of PARP1, PARP2 and PARP4 could be similar to the vault protein. The protein structure and interaction mechanisms of PARP1 and PARP2 showed similarity in catalytic domains but can differ in substrate proteins.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Till S, Diamantara K, Ladurner AG. PARP: a transferase by any other name. Nat Struct Mol Biol. 2008;15(12):1243–1244.
- Morales J, Li L, Fattah FJ, et al. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit Rev Eukaryot Gene Expr. 2014;24(1):15–28.
- Hottiger MO, Hassa PO, Luscher B, et al. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci. 2010;35(4):208–219.
- Otto H, Reche PA, Bazan F, et al. In silico characterization of the family of PARP-like Poly(ADP-ribosyl) transferases (pARTs). BMC Genomics. 2005;6(1):139.
- Meder VS, Boeglin M, de Murcia G, et al. PARP-1 and PARP-2 interact with nucleophosmin/beta 23 and accumulate in transcriptionally active nucleoli. J Cell Sci. 2005;118(1):211–222.
- Woodhouse BC, Dianov GL. Poly ADP-ribose polymerase-1: an international molecule of mystery. DNA Repair. 2008;7(7):1077–1086.
- Xu SW, Bai P, Little PJ, et al. Poly(ADP-ribose) polymerase 1 (PARP1) in atherosclerosis: from molecular mechanisms to therapeutic implications. Med Res Rev. 2014;34(3):644–675.
- Chugani DC, Rome LH, Kedersha NL. Evidence that vault ribonucleoprotein-particles localize to the nuclear-pore complex. J Cell Sci 1993;106:23–29.
- Daniel RA, Rozanska AL, Thomas HD, et al. Inhibition of poly (ADP-Ribose) polymerase-1 enhances temozolomide and topotecan activity against childhood neuroblastoma. Clin Cancer Res. 2009;15(4):1241–1249.
- Opar A. Novel anticancer strategy targets DNA repair. Nat Rev Drug Discov. 2009;8(6):437–438.
- Mangerich A, Burkle A. How to kill tumor cells with inhibitors of Poly(ADP-ribosyl)ation. Int J Cancer. 2011;128(2):251–265.
- Peralta-Leal A, Rodriguez-Vargas JM, Aguilar-Quesada R, et al. PARP inhibitors: New partners in the therapy of cancer and inflammatory diseases. Free Radical Bio Med. 2009;47(1):13–26.
- Jagtap P, Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4(5):421–440.
- Kickhoefer VA, Siva AC, Kedersha NL, et al. The 193-kD vault protein, VPARP, is a novel Poly(ADP-ribose) polymerase. J Cell Biol. 1999;146(5):917–928.
- D’Amours D, Desnoyers S, D’Silva I, et al. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342:249–268.
- Yelamos J, Farres J, Llacuna L, et al. PARP-1 and PARP-2: new players in tumour development. Am J Cancer Res. 2011;1(3):328–346.
- Yu K, Yau YH, Sinha A, et al. Modulation of the vault protein-protein interaction for tuning of molecular release. Sci Rep. 2017;7(1):1–10.
- van Zon A, Mossink MH, Schoester M, Houtsmuller AB, et al. The formation of vault-tubes: a dynamic interaction between vaults and vault PARP. J Cell Sci. 2003;116(21):4391–4400.
- González J, Gálvez A, Morales L, et al. Integrative approach for computationally inferring interactions between the alpha and beta subunits of the calcium-activated potassium channel (BK): a docking study. Bioinform Biol Insights. 2013;7:S10077–S82.
- Ünlü A. Computational prediction of actin-actin interaction. Mol Biol Rep. 2014;41(1):355–364.
- Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–1802.
- Benkert P, Tosatto SCE, Schwede T. Global and local model quality estimation at CASP8 using the scoring functions QMEAN and QMEANclust. Proteins. 2009;77(S9):173–180.
- Rissel D, Peiter E. Poly(ADP-Ribose) polymerases in plants and their human counterparts: parallels and peculiarities. IJMS. 2019;20(7):1638.
- Langelier MF, Planck JL, Roy S, et al. Crystal structures of poly(ADP-ribose) polymerase-1 (PARP-1) zinc fingers bound to DNA: structural and functional insights into DNA-dependent PARP-1 activity. J Biol Chem. 2011;286(12):10690–10701.
- Chaitanya GV, Alexander JS, Babu PP. PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun Signal. 2010;8(1):31.
- Liu L, Kong M, Gassman NR, et al. PARP1 changes from three-dimensional DNA damage searching to one-dimensional diffusion after auto-PARylation or in the presence of APE1. Nucleic Acids Res. 2017;45(22):12834–12847.
- Chen Q, Kassab MA, Dantzer F, et al. PARP2 mediates branched poly ADP-ribosylation in response to DNA damage. Nat Commun. 2018;9(1):3233.
- Vyas S, Chang P. New PARP targets for cancer therapy. Nat Rev Cancer. 2014;14(7):502–509.
- Alemasova EE, Lavrik OI. Poly (ADP-ribosyl) ation by PARP1: reaction mechanism and regulatory proteins. Nucleic acids research. 2019;47(8):3811–3827.
- Cohen M, Visochek L, Rozensal D, et al. DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: a link to histone acetylation. Mol Cell. 2007;25(2):297–308.
- Caron M-C, Sharma AK, O’Sullivan J, et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat Commun. 2019;10(1):2954.
- Borges HL, Linden R, Wang YJ. DNA damage-induced cell death. Cell Res. 2008;18(1):17–26.
- Gottschalk AJ, Timinszky G, Kong SE, et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. P Natl Acad Sci.2009;106(33):13770–13774.
- Brown JS, O'Carrigan B, Jackson SP, et al. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov. 2017;7(1):20–37.
- Bian C, Zhang C, Luo T, et al. NADP + is an endogenous PARP inhibitor in DNA damage response and tumor suppression. Nat Commun. 2019;10(1):693.