
Abstract
Daylily (Hemerocallis citrina Borani), a widespread ornamental plant around the world, has been considered as an ideal model to study flower development recently. To initiate the research to comprehensively investigate the molecular mechanism of the flower development of daylily, we performed a transcriptome sequencing study to establish the transcriptional profiles of this plant at different flower developmental stages. Briefly, we divided the flower development of daylily into six stages, and performed transcriptome sequencing for the samples collected from each stage. A total of 56,598 unique transcripts were obtained, of which 35,424 ones (about 62.6%) encode proteins with known functions based on homology searches against well-studied model plants. The expression profiles of these genes showed that 7,395 genes were differentially expressed during flower development. Further functional enrichment analysis for these differentially expressed genes revealed that they are enriched in the processes of translation, oxidation-reduction, and cell wall metabolism, suggesting the critical roles of these processes in daylily flower development.
Introduction
Daylily (Hemerocallis citrina Baroni), a popular herbaceous perennial plant, is widely cultivated around the world as an ornamental plant. Daylily has distichously arranged long, linear leaves, and showy flowers, which are borne on short pedicels [Citation1]. The flower buds are commonly used as a vegetable in Asia [Citation2]. Recent studies showed that the flowers of daylily have more antioxidant compounds than other plants, making it an excellent nutraceutical food and sparking a potential for medical use [Citation3,Citation4].
In the past few decades, cumulative studies have been performed to explore the mechanism of daylily flower development, from morphology to biochemistry, physiology and genetics. More and more evidence shows that daylily is an ideal model for plant flower development [Citation5]. Bieleski and Reid [Citation6] reported the increase of sugar and ion efflux in daylily flower senescence and they also confirmed the significant sugar content changes in daylily flower development. Panavas et al. [Citation7] reported that the ROS increase due to reduced effectiveness of protective enzymes, leads to programmed cell death (PCD), which plays important roles in daylily flower development. They also showed that up-regulation of activities of wall-based enzymes with similar functions, such as polygalacturonase and β-galactosidase, may contribute to flower senescence [Citation8]. In addition, Nitta et al. [Citation9] reported that a daylily gene, similar to TOC1 in Arabidopsis thaliana, controls flower opening; and a major locus controls the timing of flower closing. The molecular mechanism by which these two genetic loci regulate the flower development [Citation10], however, is still unclear. Here, to explore the molecular mechanism of daylily flower development from the perspective of genomics, we studied the transcriptional profiles of daylily flower at different developmental stages.
In this study, with the goal to identify essential genes involved in the regulation of daylily flower development, transcriptome sequencing was performed to compare the transcriptional profiles of six flower developmental stages. It showed that the majority of the up-regulated genes are involved in the processes of translation, oxidation-reduction and cell wall metabolism, suggesting the critical roles of these processes in flower development.
Materials and methods
Plant material and growth
Daylily plants (kindly supplied by Prof. Chen Wang at College of Life Science and Technology, Harbin Normal University), were cultivated in the experiment station of Harbin Normal University. All samples were collected on June 26th, 2019, from one daylily clone. The samples were classified into six stages based their size: stage A (2.1-2.5 cm), stage B (4.1-4.5 cm), stage C (5.1-5.5 cm), stage D (7.1-8.0 cm), stage E (full opening) and stage F (flower senescence). The details are shown in Supplemental Figure S1. Two samples were collected for each stage. All samples were flash frozen in liquid nitrogen and then stored at −80 °C.
Construction and sequencing of RNA-seq library
Total RNA was extracted using the RNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s instructions as our previous study described [Citation11]. RNA samples were sent to BGI-Shenzhen Co. Ltd (Shenzhen, China), and RNA-seq was performed on the BGI-Seq500 platform according to the manufacturer’s instructions to generate 150-bp paired-end reads as described in a previous study [Citation12].
Sequences assembly and annotation of RNA-seq data
Adapter sequences and low-quality reads were removed from raw sequences to produce clean data, which was performed by BGI Co. Ltd. These clean data were submitted to NCBI SRA database (Accession no.: SRP243199). The clean reads were then assembled de novo into contigs using Trinity with default parameters [Citation13]. The contigs were further clustered into unigenes using CORSET with default parameters as described [Citation14]. All unigenes were BLAST-searched against combined databases of Arabidopsis thaliana, rice (Oryza sativa), maize (Zea mays), orchid (Phalaenopsis equestris) and asparagus (Asparagus officinalis) protein sequences using BLASTX program for functional annotation (E-value cutoff: 1e-5) [Citation15]. Next, daylily unigenes were assigned with functional annotation based on their corresponding homologs in the combined database, including Gene Ontology (GO) and KOG (EuKaryotic Orthologous Groups) annotations [Citation16,Citation17]. In addition, daylily unigenes were scanned using the iTAK pipeline to identify transcription factors, as Jin et al. [Citation18] described.
Expression quantification and differential expression analysis
All RNA-seq reads were mapped to daylily unigenes using TopHat software [Citation19], and the expression levels of each unigene were estimated as FPKM values (fragments per kilobase of exon per million fragments mapped) using Cufflinks software [Citation20]. Differentially expressed unigenes were identified using Cufflinks software with the time-course model as the protocol described. The GO annotation results of these differentially expressed unigenes involved in flower development were retrieved, and GO functional enrichment analyses were performed using the topGO package in R [Citation21].
Results and discussion
Sequencing and de novo assembly of the daylily transcriptome
In order to establish the transcriptional profiles of daylily at different flower developmental stages, we collected two samples from each of the classified six developmental stages. RNA was extracted from each sample, and RNA-seq was performed by using next-generation sequencing. In total, around 291 million reads were generated from these twelve samples. We have submitted the data to NCBI SRA database (Accession numbers: SRP243199). After these data were de novo assembled by using Trinity software, and the redundant transcripts were eliminated using CORSET, we obtained 56,598 transcripts (genes) from these reads. The mean length of these transcripts was 1,246 bp, and the N50 value was 1,735 bp (). To determine their identities, we performed a BLAST search to compare these transcripts with the database established by combining the protein sequences of the well-studies plant organisms: Arabidopsis, rice, maize, orchid and asparagus. It showed that, of these transcripts, 35,424 ones (about 62.6%) were identified as homologs with significant hits to the well-studied plant organisms. As expected, it also showed that the longer transcripts had more hits than the short ones (), implying that the longer transcripts were more probably annotated. In other words, the identities of the longer transcripts have more confidence, which is consistent with previous reports in other plants [Citation11,Citation12].
Figure 1. Length distributions of daylily unique transcripts.
Note: The X-axis lists the length of transcripts, and Y-axis lists the numbers of transcripts. Red bars represent the total number of transcripts at specific length ranges, and the adjacent blue bars represent the ones annotated.
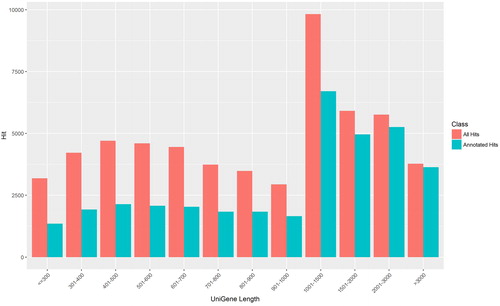
Table 1. Summary of de novo assembled the daylily transcriptome.
Functional annotation analysis of daylily unigenes
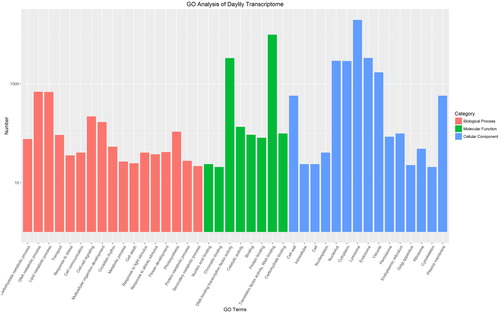
To identify the biological processes that the transcripts are involved in, we further used the GO terms and KOG annotations of their homologs to assign functions to the transcripts of daylily. It showed that 23,019 genes (about 40.67%) can be properly grouped into the following GO processes ( and Supplemental Figure S2): DNA metabolic process (GO:0006259, 699 unigenes), lipid metabolic process (GO:0006629, 689 unigenes), transport (GO:0006810, 93 unigenes), cell–cell signaling (GO:0007267, 221 unigenes), multicellular organism development (GO:0007275, 170 unigenes), circadian rhythm (GO:0007623, 54 unigenes), cell death (GO:0008219, 25 unigenes), and flower development (GO:0009908, 42 unigenes). These biological processes provide valuable information for understanding daylily flower development. For example, DNA and lipid metabolic processes imply DNA and lipids undergo tremendous changes in daylily flower development, which is consistent with previous reports [Citation4,Citation8]. Similarly, transport, cell–cell signaling, circadian rhythm and cell death are all considered critical processes in daylily flower development, which have also been reported in previous studies but with insufficient evidence. The grouping of a significant number of transcripts into the GO term “flower development” shows the significance of these genes in daylily flower development. Meanwhile, KOG annotations were also assigned to all the transcripts, and it showed that the three top KOG terms were signal transduction mechanisms (2,930 genes, 13.98%), general function prediction only (2,887 genes, 13.77%), and posttranslational modification, protein turnover and chaperones (2,171 genes, 10.35%), which are consistent with the GO annotations. In addition, plant transcript factors (TFs) were also identified among these transcripts; in total 2281 TF genes were identified and classified into 74 TF gene families according to the classification in PlantTFDB. The most abundant TF family was the MYB (166 genes), followed by the AP2/ERF (111 genes), NAC (110 genes), C2H2 (105 genes) and bHLH (104 genes) (Supplemental Figure S3). These TF families have been shown to be key in the regulation of flower development in well-studied model plants [Citation22–24], suggesting that the daylily has similar regulatory mechanisms.
Figure 2. GO annotation results of daylily unique transcripts.
Note: GO terms are listed along the X-axis, and the numbers of transcripts grouped in the GO terms are shown on the Y-axis. Red bars represent Biological processes; Green bars represent Molecular functions; and blue bars represent cellular components.
Expression analysis of genes involved in daylily flower development
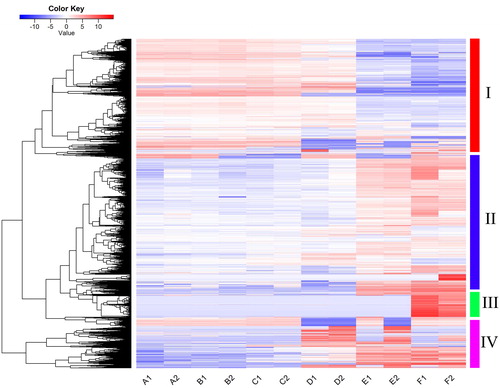
To determine which genes play a role in the daylily flower development process, five comparisons between adjacent flower development stages were performed using Cufflinks software. In total, 7,395 differentially expressed genes (DEGs) were identified with a false discovery rate of 0.01. Specifically, there were 129 genes involved in the transition from stage A to B, 348 genes involved in stage B to C, 1551 genes involved in stage C to D, 3964 genes involved in stage D to E, and 2673 genes involved in stage E to F. These genes were clustered into four groups based on their expression levels (). Group I was highly expressed at stages A, B, C and D, but less expressed at stages E and F; Group II showed the opposite trend to Group I; Group III had low expression at all stages except stage F, suggesting their special functions at stage F; whereas Group IV was highly expressed at stage D, E and F. These patterns are consistent with previous studies [Citation9], which considered flower opening and flower senescence as the most import processes during daylily flower development with the identification of the largest number of genes related to the regulation of the two processes.
Figure 3. Heatmap showing expression profiles of differentially expressed genes during daylily flower development.
Note: The heatmap was generated using R gplots package; the FPKM values of each gene were normalized and used as input. The color transition from dark blue to dark red represents the expression level of genes from the lowest to the highest.
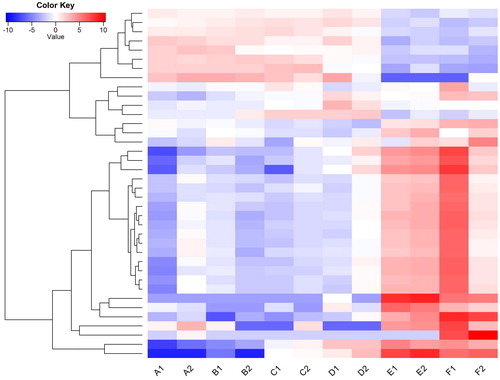
To determine the biological processes of these DEGs, GO annotation enrichment analysis was performed using the software package topGO. The analysis had 128 GO terms significantly enriched (Supplemental Table S1). It has been reported that programmed cell death (PCD) plays important role in daylily flower development [Citation25], which involves many complex biochemical pathways, including protein synthesis (translation) and proteolysis (protein disassembly). PCD is involved in cell death in flower tissues and is considered to be the major role for regulating flower development states in daylily [Citation7,Citation8,Citation25,Citation26]. Two biological processes related to PCD were identified in this study. They are cell wall metabolism and reactive oxygen species (ROS) metabolism. Our finding suggests that daylily autonomously regulates enzymes that modify cell walls, such as polygalacturonases and b-galactosidases. Also, it regulates ROS related enzymes, such as oxidoreductase. In addition, it promotes the PCD process, such as inhibition of translation, protein disassembly [Citation27], and DNA digestion [Citation28]. In the “biological processes” category, the main terms, including translation (GO:0006412), oxidation-reduction process (GO:0055114), cell wall organization or biogenesis (GO:0071554), response to auxin (GO:0009733), protein complex disassembly (GO:0043241) and response to auxin (GO:0009733), suggest that the genes involved in these processes potentially contributed to daylily flower development. These findings have well confirmed previous suggestions; for example, genes involved in cell-wall metabolism (GO:0071554) have obvious changes of expressional profiles during flower development (), implying their regulatory function in flower opening and senescence. In addition, GO enrichment analysis were enriched in terms including hydrolase activity (GO:0016798), oxidoreductase activity (GO:0016491), transporter activity (GO:0005215), ribosome (GO:0005840) and cell wall (GO:0005618) from the categories of “molecular function” and “cellular component”, which also confirmed their function in the PCD process in flower development. However, the major locus that regulates daylily flower development, as Nitta et al. [Citation9] described, is still unknown. It is reasonable to speculate that some transcription factors related to flower development regulation might be encoded by this locus.
Figure 4. Heatmap showing expression profiles of differentially expressed genes involved in cell wall metabolism during flower development.
Note: The genes that were grouped into the GO term (GO:0071554) were identified as cell–wall metabolism genes. Their expressional levels (FPKM values) were normalized, and used as input to generate the cluster plot. Color annotations are the same as those in .
Conclusions
In the present study, we performed RNA-seq to establish the transcriptional profiles of different flower developmental stages of daylily. The transcriptome sequencing data were assembled into 56,598 transcripts, and 23,019 genes were properly annotated. Importantly, 7,395 genes were identified as differentially expressed genes during daylily flower development. These genes were enriched in translation, oxidation reduction process, cell wall metabolism, and ROS metabolism, implying their critical roles in daylily flower development.
Supplemental Material
Download PDF (373.9 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Cui H, Zhang Y, Shi X, et al. The numerical classification and grading standards of daylily (Hemerocallis) flower color. Plos One. 2019;14(6):e0216460.
- Hou F, Li S, Wang J, et al. Identification and validation of reference genes for quantitative real-time PCR studies in long yellow daylily, Hemerocallis citrina Borani. PLoS One. 2017;12(3):e0174933.
- Wu W-T, Mong M-c, Yang Y-c, et al. Aqueous and ethanol etracts of daylily flower (Hemerocallis fulva L.) protect HUVE cells against high glucose. J Food Sci. 2018;83(5):1463–1469.
- Zhang Y, Cichewicz RH, Nair MG. Lipid peroxidation inhibitory compounds from daylily (Hemerocallis fulva) leaves. Life Sci. 2004;75(6):753–763.
- Rodriguez-Enriquez MJ, Grant-Downton RT. A new day dawning: Hemerocallis (daylily) as a future model organism. AoB Plants. 2013;5:pls055.
- Bieleski RL, Reid MS. Physiological changes accompanying senescence in the ephemeral daylily flower. Plant Physiol. 1992;98(3):1042–1049.
- Panavas T, Reid PD, Rubinstein B. Programmed cell death of daylily petals: Activities of wall-based enzymes and effects of heat shock. Plant Physiol Biochem. 1998;36(5):379–388.
- Panavas T, Rubinstein B. Oxidative events during programmed cell death of daylily (Hemerocallis hybrid) petals. Plant Sci. 1998;133(2):125–138.
- Nitta K, Yasumoto AA, Yahara T. Variation of flower opening and closing times in F1 and F2 hybrids of daylily (Hemerocallis fulva; Hemerocallidaceae) and nightlily (H. citrina). Am J Bot. 2010;97(2):261–267.
- Hirota SK, Nitta K, Kim Y, et al. Relative role of flower color and scent on pollinator attraction: experimental tests using F1 and F2 hybrids of daylily and nightlily. PLoS One. 2012;7(6):e39010.
- Shu Y, Li W, Zhao J, et al. Transcriptome sequencing and expression profiling of genes involved in the response to abiotic stress in Medicago ruthenica. Genet Mol Biol. 2018;41(3):638–648.
- Luo D, Zhou Q, Wu Y, et al. Full-length transcript sequencing and comparative transcriptomic analysis to evaluate the contribution of osmotic and ionic stress components towards salinity tolerance in the roots of cultivated alfalfa (Medicago sativa L.). BMC Plant Biol. 2019;19(1):32.
- Haas BJ, Papanicolaou A, Yassour M, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8(8):1494–1512.
- Davidson NM, Oshlack A. Corset: enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014;15(7):410.
- Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402.
- Tatusov RL, Fedorova ND, Jackson JD, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003 ;4:41.
- Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–29.
- Jin J, Zhang H, Kong L, et al. PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2014;42(D1):D1182–D1187.
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111.
- Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–578.
- Alexa A, Rahnenfuhrer J. topGO: enrichment analysis for gene ontology. R package version 2.38.1. 2019.
- Takahashi R, Benitez ER, Oyoo ME, et al. Nonsense mutation of an MYB transcription factor is associated with purple-blue flower color in soybean. J Hered. 2011;102(4):458–463.
- Noda K, Glover BJ, Linstead P, et al. Flower colour intensity depends on specialized cell shape controlled by a Myb-related transcription factor. Nature. 1994;369(6482):661–664.
- Liu J, Li J, Wang H, et al. Identification and expression analysis of ERF transcription factor genes in petunia during flower senescence and in response to hormone treatments. J Exp Bot. 2011;62(2):825–840.
- van Doorn WG, Woltering EJ. Senescence and programmed cell death: substance or semantics? J Exp Bot. 2004;55(406):2147–2153.
- Van Hautegem T, Waters AJ, Goodrich J, et al. Only in dying, life: programmed cell death during plant development. Trends Plant Sci. 2015;20(2):102–113.
- Stephenson P, Rubinstein B. Characterization of proteolytic activity during senescence in daylilies. Physiol Plant. 1998;104(3):463–473.
- Panavas T, LeVangie R, Mistler J, et al. Activities of nucleases in senescing daylily petals. Plant Physiol Biochem. 2000;38(11):837–843.