
Abstract
Euphorbia kansui Liou is an endemic and traditional medicinal plant in China. Despite its value, the genomic and transcriptomic information of E. kansui is limited. In the present study, a total of 43,211,690 high-quality reads were generated using Illumina paired-end sequencing technology. After assembly, 58,362 unigenes were recovered with an N50 length of 1,683 bp. 36,396 (62.36%) of these unigenes had at least one blast hit against the multiple public databases. Based on KEGG pathway analyses, unigenes were discovered to encode known enzymes that catalyze the formation of the terpenoid backone and diterpenoid. In addition, the unigenes related to casbene synthase, which catalyzes the formation of the major active constituents (diterpenoids) in E. kansui, were identified. For functional marker development, 7,016 candidate simple sequence repeats (SSR, microsatellites) were identified from 6,150 unigenes, and 40 SSR loci were selected randomly and amplified in two populations of E. kansui. A total of 28 loci were amplified successfully and 23 loci exhibited polymorphisms. The number of alleles of each locus ranged from 2 to 8 alleles and the mean number of alleles per locus was 3.391. The observed heterozygosity and expected heterozygosity varied from 0.100 to 1.000 and 0.099 to 0.809, respectively. The microsatellite markers developed here will be useful for studies on population genetics of this endemic species. Moreover, the information of the transcriptome will provide a useful resource for the future research on metabolism, regulation of the terpenoid biosynthesis, genetic and proteome studies of E. kansui.
Introduction
Euphorbia kansui Liou is a perennial herb of Euphorbia (Euphorbiaceae). It is an endemic species of China, and is distributed mainly in Shaanxi, Henan and Gansu provinces. The dried root of E. kansui is a commonly used and well-known traditional Chinese medicine and is listed in the Chinese Pharmacopoeia. It is widely applied to treat with edema, ascites, schistosomiasis and asthma [Citation1]. It was reported that E. kansui showed curative effects on anti-tumor [Citation2], anti-viral [Citation3], inhibition cell division [Citation4, Citation5], anti-fertility [Citation6] and pesticidal activity [Citation7]. At present, it is often used clinically to treat pancreatitis, intestinal obstruction, hydrothorax, ascitic fluid phlegm syndrome and cirrhotic ascites. In recent years, the genome sequences of some plants in Euphorbiaceae have been completed, such as Ricinus communis [Citation8], Jatropha carcas [Citation9] and Hevea brasiliensis [Citation10]. However, genomic information about E. kansui is lacking, and the current lack of genomic sequence data limits the molecular research. In our previous study, we reported the development and ultrastructure of laticifers and the latex proteome of E. kansui, but identifying proteins involved in the development of laticifers has been challenging greatly because of lack of its genome and transcriptome information [Citation11].
Transcriptome is the set of transcripts of an organism at a defined spatial and temporal locus, which provides valuable and comprehensive information on gene expression, gene regulation, and amino acid content of proteins [Citation12]. In recent years, next-generation sequencing (NGS) technologies, such as Illumina paired-end transcriptome analysis, provide new opportunities to acquire transcriptomic data for the purpose of gene mining and molecular marker development, especially for species without a reference genome [Citation13]. Wang et al. (2009) reported the transcriptome data of Artemisia annua, and characterized the terpenoid biosynthesis pathway genes, sesquiterpene synthase and monoterpene synthase [Citation14]. The candidate genes associated with carotenoid biosynthesis in Lycium chinense were discovered and further confirmed based on transcriptome data [Citation15].
Microsatellites, also called simple sequence repeats (SSR), are found in the nuclear genomes of all eukaryotes and are often abundant and evenly dispersed [Citation16]. SSR markers are highly informative and widely used for evolution and genetics studies [Citation17]. Transcriptome sequencing is capable of identifying massive numbers of potential SSR [Citation18]. Illumina paired-end sequencing was successfully used for developing SSR markers of non-model organisms, including Mucuna prurien [Citation19], Dendrobium officinale [Citation20]. In E. kansui, only 12 genome SSR markers were developed [Citation21], but no EST-SSR markers are available to date. EST-SSR markers predicted and developed in this study will enrich the number of molecular markers, and facilitate gene mapping, linkage map development and genetic diversity analysis of this endemic species.
In the present study, we performed large-scale transcriptome sequencing using Illumina paired-end sequencing technology to obtain comprehensive genomic information. SSR markers were developed based on the transcriptome sequences for genetic diversity studies of E. kansui. After annotation, we analyzed candidate genes involved in terpenoids biosynthesis. To our knowledge, this is the first systematic report about transcriptome analysis of E. kansui. This study will provide the basis for molecular biology, genetics and further proteomic research of E. kansui.
Materials and methods
Plant material
Euphorbia kansui plants were obtained from the field at the Botanical Garden of Northwest University (34.25 N, 108.92 E), Xi’an Botanical Garden (34.21 N, 108.95 E) and Yongshou county (34.70 N, 108.15 E) in Shaanxi Province (Shaanxi, People’s Republic of China). Tender leaves, inflorescence and fruits were collected and flash frozen in liquid nitrogen and stored at −80 °C until RNA extraction.
The leaves of 20 individuals (10 individuals of each population) were sampled from the Botanical Garden of Northwest University (34.25 N, 108.92 E) and Xi’an Botanical Garden (34.21 N, 108.95 E) respectively to develop SSR markers. These plants were transplanted from different wild populations.
RNA extraction
To globally characterize the E. kansui transcriptome and enhance sequence coverage, the total RNA was isolated respectively from young leaves, inflorescence and fruits using RNA extraction Trizol Kit (Gibco BRL, Grand Island, USA) following the manufacturer’s instructions. Then the total RNA was treated with RNase-free DNase I (Takara Bio, Japan) for 30 min at 37 °C to remove residual DNA. The quantity and quality of the isolated RNA were assessed by Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Equal amounts of RNA isolated from different tissues were mixed for the subsequent cDNA library construction step.
cDNA library construction and illumina sequencing
Transcriptome library for sequencing was constructed according to the Illumina TruSeq RNA library protocol outlined in “TruSeq RNA Sample Preparation Guide” (Illumina 2009). Briefly, poly(A) mRNA was purified using Sera-mag Magnetic Oligo (dT) Beads (part of the mRNA-seq Preparation kit, Illumina, USA). The purified mRNA was first chopped into short fragments by adding fragmentation buffer, and the cleaved RNA fragments were reverse transcribed into first-strand cDNA with reverse transcriptase and random hexamer primers. Then second-strand cDNA was synthesized by using DNA polymerase I, dNTPs and RNase H. The short double-stranded cDNA fragments were purified using a QiaQuick PCR extraction kit (Qiagen, Hilden, Germany), and these purified fragments was then washed with EB buffer for end repair and A tailing. Then, Illumina Adapters were ligated to sequencing adapters using T4 DNA ligase. Following agarose gel electrophoresis and extraction of cDNA from gels, the cDNA fragments were purified and enriched by PCR to construct the final cDNA library. Finally, the cDNA library was sequenced on the Illumina sequencing platform (Illumina HiSeq TM 2000 system) using the paired-end technology in a single run.
De novo assembly and data analysis
The raw data from the images acquired after sequencing was transformed by base-calling into raw reads and stored in FASTQ format. Clean reads were selected by removing reads containing adaptor sequences, reads with more than 5% N base (base unknown) low quality sequences. And the clean reads were assembled using Trinity and TGICL to construct unique consensus sequences with the default setting.
Gene annotation
All obtained unigenes were annotated with the basic local alignment search tool (BLASTx) analysis with an E-value cut-off of 10−5 against the databases including non-redundant protein (Nr) database, Swiss-Prot protein database, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database, and the Cluster of Orthologous Groups (COG) database to retrieve proteins with the highest sequence similarity score with the given unigenes along with their protein functional annotations. COG database was used to analyze, predict and classify unigenes with probable functions, whereas KEGG database was used to assign pathways of unigenes. The Blast2 GO program was used to obtain the annotation result of all the unigenes in Gene Ontology (GO) database which can describe the information of genes through three ontologies: molecular function, biological process and cellular component. After that, statistical analysis of GO functional classification was performed by WEGO software.
Identification of SSR loci, primer design and marker validation
SSR markers were detected using MIcroSAtellite tool (http://pgrc.ipk-gatersleben.de/misa/). SSRs were considered to contain motifs with two to six nucleotides in size and a minimum of 4 contiguous repeat units. Then primer pairs were designed by primer 3.0 (http://www.broadinstitute.org/genome_software/other/primer3.html) in the flanking regions of SSRs. Primers were designed based on the following criteria: GC content ranging from 40% to 60%, primer length ranging from 18 to 27 bp, melting temperature ranging from 55 to 63 °C and the expected PCR product sizes ranging from 100 to 280 bp.
A total of 40 primer pairs were randomly selected and synthesized by Sangon biotechnology Co., Ltd (Shanghai, China). For validation of SSR primers, total genomic DNA was extracted from young leaves of 20 individuals from two populations described previously (ten for each one) using plant genomic DNA kit (Tiangen, Beijing, China). PCR amplifications were performed in a total volume of 10 μL containing 1 ng/μL DNA, 5 μL 2 × Taq PCR master mix (0.1 U Taq polymerase/μL; 500 µmol/L each dNTP; 20 mmol/L Tris–HCl, pH8.3; 100 mmol/L KCl; 3.0 mmol/L MgCl2) and 0.2 μmol/L of each forward and reverse primer and 3.6 μL ddH2O. The PCR amplification was performed under the following conditions: initial denaturation at 94 °C for 5 min followed by 30 cycles of 94 °C for 50 s, 30 s annealing at 55–64 °C, 45 s extension at 72 °C, and 5 min at 72 °C for final extension. The PCR products were separated by 10% polyacrylamide gel electrophoresis; then the gels were stained with silver nitrate. Fragment sizes of each locus were estimated using Quantity One Software (Bio-Rad Laboratories, Drive Hercules, CA, USA) with pBR322 DNA/MspI as the molecular weight standard (Tiangen, Beijing, China).
Microsatellites data analysis
The number of alleles (NA), expected heterozygosity (HE), observed heterozygosity (HO), polymorphic information content (PIC) and Hardy-Weinberg (HW) equilibrium were calculated using CERVUS version 3.0.
Results and discussion
Sequence assembly and data analysis
The next-generation sequencing technology has provided a powerful and cost-efficient tool to obtain large amounts of transcriptome data from many organisms and tissue types without a reference genome [Citation22, Citation23]. In the present study, RNA was extracted from fresh leaves, inflorescence and fruits. The extracted RNA from these tissues was mixed equally and sequenced with Illumina paired-end sequencing technology, so as to find more unigenes of E. kansui. A similar method has been used in transcriptome sequencing of Benincasa hispida [Citation18] and Camelia sativa [Citation24]. After cleaning the adaptor sequences and quality checks, about 43,211,690 clean reads were obtained amounting to 4,321,169,000 base pairs (bp), and 97.80% of Q20 bases, and these reads had an average GC content of 44.09% (). The raw data were deposited in the NCBI Sequence Read Archive (SRA) database (accession number: SRP067381).
Table 1. Summary of transcriptome sequencing dataset.
After length and quality-based filtering, a total of 61,712 contigs were generated, containing 5,61,76,504 bp total nucleotides, with an average length of 910 bp, and a N50 of 1,530 bp (). The contigs with the length more than 500 bp accounted for about 53.18% (32,817), and 2,012 (3.26%) contigs were longer than 3,000 bp (; ). Then, de novo assembly of these contigs generated 58,362 unigenes which did not contain gap regions. Both the N50 and average length of the unigenes were 1,683 bp (). The mean length was longer than the results shown in previous studies, such as Erigeron breviscapus (916 bp) [Citation25] and Litchi chinesis (710 bp) [Citation26]. N50 is a value to evaluate the assembly quality, and the N50 length was longer than those in other species [Citation18, Citation22]. These results suggested that the transcriptome sequencing data were effectively assembled and it is possible to obtain reliable unigene sequences. Meanwhile, these data also indicated that Illumina paired-end sequencing technology is useful for the de novo sequencing and assembly of transcriptomes of non-model plants. The length of assembled unigenes ranged from 201 bp to 12,178 bp. The length distribution of the unigenes showed that 28,224 (48.36%) were 200–500 bp long, 30,138 (51.64%) unigenes were longer than 500 bp, and those with a length of over 3,000 bp were 1,786 (3.06%) (; ). A total of 23,805 unigenes (40.79%) were realigned with 11–100 reads, which comprised the largest group; 14,473 (24.80%) and 4949 (8.48%) unigenes were remapped by more than 100 and 1000 reads, respectively; whereas only 273 (0.47%) unigenes were remapped with more than 10,000 reads ().
Figure 1. Assembly length distribution of contigs and unigenes in Euphorbia kansui.
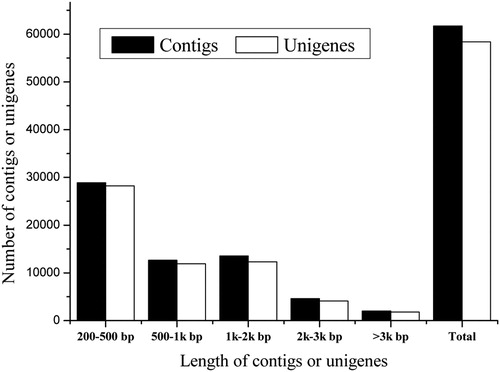
Figure 2. Number of unigenes annonated in protein databases by BLASTx with E-value threshold of 10−5. The numbers in the circles indicate the number of unigenes annotated by single or multiple databases.
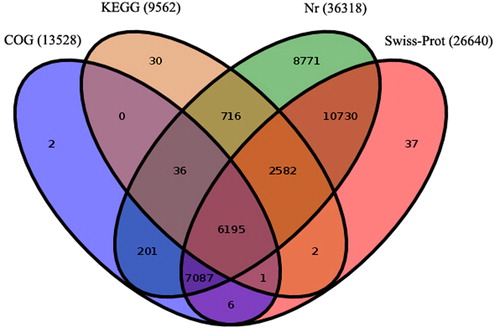
Figure 3. Gene ontology classification of assembled unigenes of Euphorbia kansui. In total 15,506 unigenes were categorized into three main categories: biological process, cellular component and molecular function.
Functional annotation of unigenes
All the unigenes were searched using BLASTx against Nr, Swiss-Prot, COG and KEGG databases with an E-value threshold of 10−5. The results suggested that among the 58,362 unigenes, a total of 36,396 (62.36%) unigenes were annotated in the public databases. 36,318 (62.23%) unigenes and 26,640 (45.65%) unigenes were homologous with the Nr database and Swiss-Prot database respectively (). Among them, 6195 unigenes were annotated in all four databases and 8771 unigenes were annotated uniquely in Nr database, 37 uniquely in Swiss-Prot database, 2 unigenes in COG database and 30 unigenes uniquely in KEGG database (). This implied that the transcriptome sequencing yielded a great number of unique genes, while 21,966 (37.64%) unigenes were not annotated in part because the whole genome sequence data was not available for E. kansui. The unigenes that had no hits to any sequences in the public databases may represent novel genes [Citation27]. We also analyzed the species distribution of BLAST hits for each unigene in the Nr database. The species distribution showed that 9990 (27.51%) of the mapped unigenes were annotated by Theobroma cacao homologs, 5252 (14.46%) from Vitis vinifera, and 2531 (6.97%) from the Fragaria vesca subsp. vesca ().
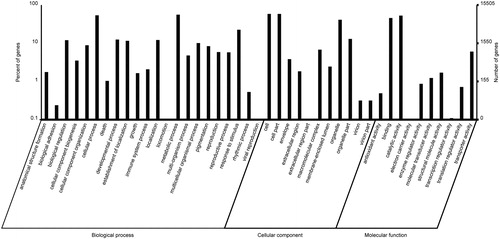
Figure 4. Cluster of Orthologous Groups (COG) function classification of Euphorbia kansui unigenes. A total of 13,528 unigenes were annotated and classified into 25 different classifications.
Gene ontology classification and COG classification
In this study, 15,506 (42.70%) unigenes were assigned with at least one GO term under the three main categories: biological process, cellular component and molecular function (). The functionally assigned unigenes covered 43 GO categories. Under biological process, 8635 (55.69%) unigenes were assigned to metabolic process, followed by cellular process (8341, 53.79%) and response to stimulus (3484, 22.47%), whereas only a few unigenes were assigned to locomotion (8, 0.05%) and viral reproduction (13, 0.08%) (). For the cellular component class, the cell (9037, 58.28%) and cell part (9037, 58.28%) were the dominant terms, whereas the least abundant term was extracellular region part (11, 0.07%) (). For the molecular function category, the major terms were catalytic activity (8,159, 52.62%) and binding (7,069, 45.59%), whereas the least abundant terms were electron carrier activity (15, 0.10%) and transcription regulator activity (17, 0.11%) ().
All unigenes obtained in our study were searched against the COG database for functional prediction and classification. Overall, 13,528 unigenes were annotated and assigned to 25 COG categories ( and Citation4). The cluster of general function prediction represented the largest one (4,552, 33.65%), followed by replication, recombination and repair (2,388, 17.65%), transcription (2340, 17.30%), posttranslational modification, protein turnover, chaperones (1,853, 13.70%), signal transduction mechanisms (1,776, 13.13%), translation, ribosomal structure and biogenesis (1,625, 12.01%), carbohydrate transport and metabolism (1,516, 11.21%), function unknown (1,439, 10.64%) and cell wall/membrane/envelope biogenesis (1,119, 8.27%), whereas only a few unigenes were assigned to extracellular structures (9, 0.07%) and nuclear structure (6, 0.04%) (). A large number of unigenes were assigned to 43 GO categories and 25 COG classifications, which indicated that our transcriptome data represented a broad diversity of transcripts in E. kansui genome. These annotations provide comprehensive information on the unigene functions.
Function classification by KEGG
All the unigenes were analyzed by the KEGG pathway database to further identify the active biochemical pathways. In total, 9,562 unigenes were mapped to 269 KEGG pathways, and some unigenes were assigned to more than one KEGG pathway. All the KEGG pathways are listed in . Among the 9,562 unigenes, 420 (4.39%) were assigned to spliceosome, which was the largest group among the KEGG categories. The other large pathway groups were ribosome (365, 3.82%), protein processing in endoplasmic reticulum (289, 3.02%) and RNA transport (281, 2.94%). In particular, unigenes involved in terpenoid backone biosynthesis (68 unigenes), diterpenoid biosynthesis (22 unigenes), fatty acid metabolism (54 unigenes) and flavonoid biosynthesis (27 unigenes) were all identified in the database ().
Identification of candidate genes involved in terpenoid biosynthesis
Diterpenoids are major constituents of E. kansui [Citation28]. The key genes involved in terpenoid backbone (isopentenyl diphosphate) and diterpenoid biosynthesis pathways were discovered, such as the genes encoding thiolase, 3-hydroxy-3-methylglutaryl coenzyme A synthase, mevalonate disphosphate decarboxylase and so on (, ). 3-Hydroxy-3-methylglutaryl-CoA reductase (HMG-CoA reductase) is a key rate-limiting enzyme in the mevalonate (MVA) pathway, which catalyzes the conversion of HMG-CoA into mevalonate. Genes that encode HMG-CoA reductase have been cloned and characterized from E. pekinensis and Hevea brasiliensis [Citation29, Citation30]. Mevalonate disphosphate decarboxylase was identified in latex of E. helioscopia and E. kansui. Whereafter, mevalonate disphosphate decarboxylase gene was cloned and it was specifically expressed in different tissues in E. helioscopia [Citation31, Citation32]. In addition, three downstream enzymes (geranyl-diphosphate synthase, geranylgeranyl pyrophosphate synthase and farnesyl pyrophosphate synthase) involved in terpenoid biosynthesis were also discovered in the transcriptome data.
Figure 5. Terpenoid backbone and diterpene biosynthesis pathways. Terpenoid backbone (isopentenyl diphosphate) is synthesized from two distinct pathways, methylerythritol phosphate (MEP/DXP) pathway and mevalonate (MVA) pathway. The enzymes participating in each step of these pathways are listed in .
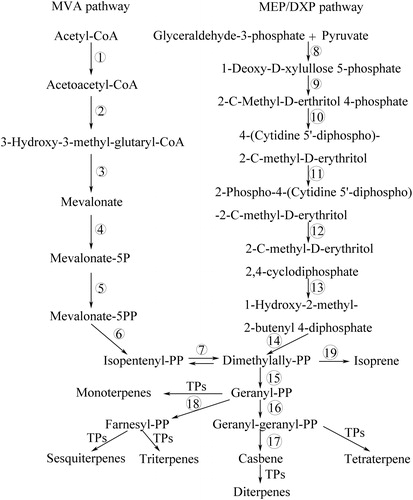
Table 2. Identified unigenes of terpenoid backbone and diterpene biosynthesis.
We also identified the candidate genes which encoded casbene synthase. In Euphorbiaceae, casbene synthase (diterpene synthase) is a key enzyme in the diterpenens biosynthesis pathway. It catalyzes the synthesis of macrocyclic diterpene olefins casbene [Citation33]. Kirby et al. (2010) discovered genes encoding putative casbene synthases in Homalanthus nutans, Sapium sebiferum, E. resinifera, E. esula and R. communis, and they considered that casbene is likely the precursor to a large number of Euphorbiaceae diterpenes [Citation34]. Casbene synthase has been isolated and characterized, and its cDNA was cloned from R. communis [Citation35, Citation36] and roots of E. fischeriana [Citation37]. The information about the unigene sequences in the transcriptome data provides a vital resource for cloning the cDNA of enzymes, and will facilitate the future research of terpenoid biosynthesis and regulation in E. kansui.
Est-SSR discovery and validation
Microsatellite markers have numerous uses, including linkage map development, quantitative trait loci mapping, marker-assisted selection, parentage analysis, cultivar fingerprinting, genetic diversity studies, gene flow and evolutionary studies [Citation38]. Recently, transcriptome sequencing has become a viable method for microsatellite mining and SSR marker development, and has been utilized in many species [Citation39]. Traditional strategies for SSR marker development are labour-intensive and costly. In E. kansui, 12 genome SSR markers were developed using paired-end Illumina shotgun sequencing [Citation21]. However, no EST-SSR markers were reported. The lack of molecular markers has impeded the genetic analysis of E. kansui, an important medicinal plant. the In the present study, all the 58,362 unigenes generated were used to mine potential simple sequence repeats, which were defined as di- to hexa-nucleotide SSR with a minimum of four repeats for all motifs (except for di-nucleotide with a minimum of six repeats, and tri-nucleotide with a minimum of five repeats). A total of 7,016 SSR loci were detected in 6,150 unigenes, out of which, 747 unigenes contained more than one SSR (). The SSRs were highly distributed over the sequences, providing a useful resource for further research of these SSR loci as genetic markers. The most abundant motif of EST-SSR varied with species. Detailed analysis showed that tri-nucleotide repeat motifs were the most abundant types with a frequency of 58.87%, and this finding was consistent with previous reports of Camelina sativa (38.21%) [Citation24] and Chrysanthemum indicum (52.32%) [Citation40], followed by di-nucleotide (25.06%), tetra-nucleotide (9.78%), penta-nucleotide (4.06%) and hexa-nucleotide repeat motifs (2.24%) (). The frequencies of SSR distributed in different repeat numbers are shown in . SSRs with five tandem repeats were the most abundant (2384), followed by those with six tandem repeats (1900), seven tandem repeats (1072) and four tandem repeats (886). Motifs that showed more than 12 repeats (di-nucleotide, 9; tri-nucleotide, 1; hexa-nucleotide, 1) were very rare. The most abundant motifs were tri-nucleotide repeat units AAG/CTT, ATC/ATG and AAT/ATT with frequencies of 24.10%, 10.46% and 6.74%, respectively. Among the di-nucleotide repeat units, AG/CT (18.87%) and AT/AT (4.36%) were the most abundant motifs (). This showed that AAG/CTT and AG/CT were the most common motifs. Similar findings have also been reported in Manihot esculenta [Citation41] and Mentha piperita [Citation42].
Figure 6. Distribution of simple sequence repeats (SSR) in Euphorbia kansui: distribution of different repeat types (a); number of SSRs per different tandem repeat motifs (b).
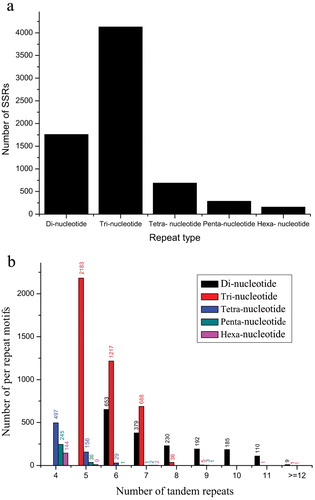
Figure 7. Frequency distribution of main SSR repeat motifs.
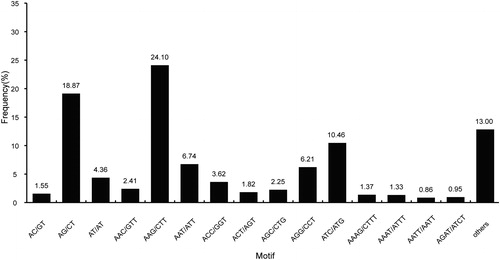
Table 3. Summary of SSR search results.
A total of 4,146 SSR loci were designed by primer 3.0, and detailed information of the SSRs discovered in this study is described in . A total of 40 loci (di-, tri-, tetra-, penta- and hex-anucleotide repeats) were randomly selected for primer synthesis and validation among two populations with 20 E. kansui individuals. Among the 40 primer pairs, 28 (70.00%) successfully yielded amplification products, whereas the other 12 SSR loci had no amplified products at the expected size (). Among them, 23 primers could be easily and consistently amplified and were polymorphic in two populations. The number of alleles (NA) of each locus ranged from 2 to 8 alleles with an average of 3.391. The polymorphic information content (PIC) ranged from 0.094 to 0.758 with an average of 0.468. The observed heterozygosity (HO) and expected heterozygosity (HE) varied from 0.100 to 1.000 and 0.099 to 0.809, with an average of 0.735 and 0.550, respectively (). In 23 polymorphic EST-SSRs, E27704, E4746, E57686 and E58997 showed significant departures from the Hardy-Weinberg (HW) equilibrium (). Five loci were monomorphic (), and those SSRs had no detectable polymorphism within two populations of E. kansui. We speculated that it is possibly because of the small sample size and population (two populations). Possibly, the SSRs exhibited polymorphism among other populations which were distributed in other regions. The other 12 loci did not amplify products at the expected size, perhaps because of the presence of introns in the unigenes, or because there may have been some errors in the base calls or assembly that resulted in incorrect flanking sequences from which primers were designed [Citation43, Citation44].
Table 4. Characterization of 23 polymonophic SSR markers in Euphorbia kansui.
EST-SSRs derived from transcribed regions are highly conserved [Citation45]. EST-SSRs have an enhanced cross-species transferability and efficiency of amplification in comparison with SSRs located in non-transcribed regions [Citation46]. Transferability of EST-SSRs has been reported in Euphorbiaceae plants including Hevea brasiliensis and Jatropha curcas. The cross-species transferability analysis has become a very popular strategy to the development of SSR markers in closely related species [Citation47, Citation48]. As a result, the developed 23 polymorphic EST-SSR markers and potential SSR loci will likely be valuable for genetic analyses of E. kansui and other close Euphorbia species.
Conclusions
In this study, we used high-throughput sequencing data to characterize the transcriptome of Euphorbia kansui, a species for which no genomic data are available. The candidate genes involved in terpenoid biosynthesis were identified. A total of 7,016 SSR loci were detected based on the transcriptome sequences. Forty ones were validated and 23 ones were polymorphic. Transcriptome data and the developed SSR markers provide a valuable resource for medicinal ingredient biosynthesis and genetic studies in E. kansui and other closely related Euphorbia species.
Supplemental Material
Download MS Excel (1.1 MB)Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
Related Research Data
References
- Chinese Pharmacopeia Committee. Phamacopoeia of the People’s Republic of China. Beijing: Chinese Medical Science Press, 2010, 81.
- Whelan LC, Ryan MF. Ethanolic extracts of Euphorbia and other ethnobotanical species as inhibitors of human tumour cell growth. Phytomedicine. 2003;10(1):53–58.
- Zheng WF. Study on in vivo antiviral activity of four diterpenoids from ethanol extracts of Euphorbia kansui. Chin Tradi Herbal Drugs. 2004;35:65–68.
- Wang LY, Wang NL, Yao XS, et al. Diterpenes from the roots of Euphorbia kansui and their in vitro effects on the cell division of Xenopus. J Nat Prod. 2002;65(9):1246–1251.
- Wang L-Y, Wang N-L, Yao X-S, et al. Euphane and tirucallane triterpenes from the roots of Euphorbia kansui and their in vitro effects on the cell division of Xenopus. J Nat Prod. 2003;66(5):630–633.,
- Han XY. The clinical research of Kansui roots and its anti-fertility effects. J Med Res. 1980;5:8–11.
- Dang QL, Choi YH, Choi GJ, et al. Pesticidal activity of ingenane diterpenes isolated from Euphorbia kansui against Nilaparvata lugens and Tetranychus urticae. J Asia-Pac Entomol. 2010;13(1):51–54.
- Chan AP, Crabtree J, Zhao Q, et al Draft genome sequence of the oilseed species Ricinus communis. Nat Biotechnol. 2010;28(9):951–956.
- Sato S, Hirakawa H, Isobe S, et al. Sequence analysis of the genome of an oil-bearing tree, Jatropha curcas L. DNA Res. 2011;18(1):65–76.
- Rahman AY, Usharraj AO, Misra BB, et al. Draft genome sequence of the rubber tree Hevea brasiliensis. BMC Genomics. 2013;14:75. [23375136]
- Zhao XY, Si JJ, Miao Y, et al. Comparative proteomics of Euphorbia kansui Liou milky sap at two different developmental stages. Plant Physiol Biochem. 2014;79:60–65.
- Morozova O, Marra MA. Applications of next-generation sequencing technologies in functional genomics. Genomics. 2008;92(5):255–264.
- Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–652.
- Wang W, Wang Y, Zhang Q, et al. Global characterization of Artemisia annua glandular trichome transcriptome using 454 pyrosequencing. BMC Genomics. 2009;10:465 [cited 2019 Dec 10]
- Wang G, Du XL, Ji J, et al. De novo characterization of the Lycium chinense Mill. leaf transcriptome and analysis of candidate genes involved in carotenoid biosynthesis. Gene. 2015;555(2):458–463.
- Tautz D, Renz M. Simple sequences are ubiquitous repetitive components of eukaryotic genomes. Nucleic Acids Res. 1984;12(10):4127–4138.
- Ellegren H. Microsatellites: simple sequences with complex evolution. Nat Rev Genet. 2004;5(6):435–445.
- Jiang B, Xie DS, Liu WR, et al De novo assembly and characterization of the transcriptome, and development of SSR markers in Wax Gourd (Benicasa hispida)). PLoS One. 2013;8(8):e71054.
- Sathyanarayana N, Pittala RK, Tripathi PK, et al. Transcriptomic resources for the medicinal Legume Mucuna pruriens: de novo transcriptome assembly, annotation, identification and validation of EST-SSR markers. BMC Genomics. 2017;18(1):409.
- Xu M, Liu X, Wang J, et al. Transcriptome sequencing and development of novel genic SSR markers for Dendrobium officinale. Mol Breeding. 2017;37(2):18.
- Yan XH, Fang MF, Qian ZQ, et al. Isolation and characterization of polymorphic microsatellites in the perennial herb Euphorbia kansui using paired-end Illumina shotgun sequencing. Conserv Genet Resour. 2014;6(4):841–843.
- Chen C, Xu M, Wang C, et al. Characterization of the Lycium barbarum fruit transcriptome and development of EST-SSR markers. PLoS One. 2017;12(11):e0187738.
- He CT, Li ZL, Zhou Q, et al. Transcriptome profiling reveals specific patterns of paclitaxel synthesis in a new Taxus yunnanensis cultivar. Plant Physiol Biochem. 2018;122:10–18.
- Mudalkar S, Golla R, Ghatty S, et al De novo transcriptome analysis of an imminent biofuel crop, Camelina sativa L. using Illumina GAIIX sequencing platform and identification of SSR markers. Plant Mol Biol. 2014;84(1-2):159–171.
- Jiang NH, Zhang GH, Zhang JJ, et al. Analysis of the transcriptome of Erigeron breviscapus uncovers putative scutellarin and chlorogenic acids biosynthetic genes and genetic markers. PLoS One. 2014;9(6):e100357.
- Lu XY, Kim H, Zhong SL, et al. De novo transcriptome assembly for rudimentary leaves in Litchi chinesis Sonn. and identification of differentially expressed genes in response to reactive oxygen species. BMC Genomics. 2014;15:805.
- Chen XM, Li JK, Xiao SJ, et al. De novo assembly and characterization of foot transcriptome and microsatellite marker development for Paphia textile. Gene. 2016;576(1 Pt 3):537–543.
- Li YL, Yuan D, Xu X, et al. Studies on macrocyclic jatrophan diterpenes of Euphorbia kansui. Chin J Chin Mate Med. 2008;33:1836–1839.
- Cao XY, Jiang JH, Liu Q, et al. Cloning and analysis of a cDNA encoding key enzyme gene (hmgr) of the MVA pathway in medicinal plant Euphorbia pekinensis. J Wuhan Bot Res. 2007;25:123–126.
- Sando T, Takaoka C, Mukai Y, et al. Cloning and characterization of mevalonate pathway genes in a natural rubber producing plant, Hevea brasiliensis. Biosci Biotech Bioch. 2008;72:2019–2060.
- Chai J, Wang D, Peng Y, et al. Molecular cloning, expression and immunolocalization analysis of diphosphomevalonate decarboxylase involved in terpenoid biosynthesis from Euphorbia helioscopia L. Biotechnol Biotechnol Equip. 2017;31(6):1106–1115.
- Zhao XY, Zhang Y, Wang M, et al Comparative proteomic analysis of latex from Euphorbia kansui laticifers at different development stages with and without UV-B treatment via iTRAQ-coupled two-dimensional liquid chromatography-MS/MS. Funct Plant Biol. 2019;47(1):67–79.
- He YF, Gao W, Liu TS, et al. Research advances of diterpene synthase. Acta Pharm Sinica. 2011;46:1019–1025.
- Kirby J, Nishimoto M, Park JG, et al. Cloning of casbene and neocembrene synthases from Euphorbiaceae plants and expression in Saccharomyces cerevisiae. Phytochemistry. 2010;71(13):1466–1473.
- Dueber MT, Adolf W, West CA. Biosynthesis of the diterpene phytoalexin casbene: partial purification and characterization of casbene synthetase from Ricinis communis. Plant Physiol. 1978;62(4):598–603.
- Mau CJ, West CA. Cloning of casbene synthase cDNA: evidence for conserved structural features among terpenoid cyclases in plants. Proc Natl Acad Sci USA. 1994;91(18):8497–8501.
- Liu HW, Tang Q, Yang YF, et al. Casbene synthase gene cloned from Euphorbia fischeriana Steud. Mol Plant Breed. 2012;10:62–66.
- Zalapa JE, Cuevas H, Zhu H, et al. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am J Bot. 2012;99(2):193–208.
- Xing W, Liao J, Cai M, et al. De novo assembly of transcriptome from Rhododendron latoucheae Franch. using Illumina sequencing and development of new EST-SSR markers for genetic diversity analysis in Rhododendron. Tree Genet Genomes. 2017;13:53.
- Han Z, Ma X, Wei M, et al. SSR marker development and intraspecific genetic divergence exploration of Chrysanthemum indicum based on transcriptome analysis. BMC Genomics. 2018;19(1):291
- Raji AA, Anderson JV, Kolade OA, et al. Gene-based microsatellites for cassava (Manihot esculenta Crantz): prevalence, polymorphisms, and cross-taxa utility. BMC Plant Biol. 2009;9:118.
- Kumar B, Kumar U, Yadav HK. Identification of EST-SSRs and molecular diversity analysis in Mentha piperita. Crop J. 2015;3(4):335–342.
- Torales SL, Rivarola M, Pomponio MF, et al. De novo assembly and characterization of leaf transcriptome for the development of functional molecular markers of the extremophile multipurpose tree species Prosopis alba. BMC Genomics. 2013;14:705
- Hu Z, Zhang T, Gao XX, et al. De novo assembly and characterization of the leaf, bud, and fruit transcriptome from the vulnerable tree Juglans mandshurica for the development of 20 new microsatellite markers using Illumina sequencing. Mol Genet Genomics. 2016;291(2):849–862.
- Varshney PK, Graner A, Sorrells ME. Genic microsatellite markers in plants: features and applications. Trends Biotechnol. 2005;23(1):48–55.
- Barbara T, Palma-Silva C, Paggi GM, et al. Cross-species transfer of nuclear microsatellite markers: potential and limitations. Mol Ecol. 2007;16(18):3759–3767.
- Yan Z, Wu F, Luo K, et al. Cross-species transferability of EST-SSR markers developed from the transcriptome of Melilotus and their application to population genetics research. Sci Rep. 2017;7(1):59–79.
- Yadav HK, Ranjan A, Asif MH, et al. EST-derived SSR markers in Jatropha curcas development, characterization, polymorphism, and transferability across the species/genera. Tree Genet Genomes. 2011;7(1):207–219.