
Abstract
Red-fleshed pummelo (Citrus grandis), a mutant of ‘Guanxi’, is an important economic plant rich in carotenoids. The transcriptome of ‘Guanxi’ pummelo was compared with red-fleshed pummelo, with the aim of identifying the key genes responsible for red flesh through RNA sequencing. In total, 44.11 and 47.71 Gb clean reads were respectively obtained after filtering in red and white flesh. Altogether 46,425 unigenes, with an average length of 1208 bp, were assembled. Compared with that of white flesh, 3722 genes were up-regulated, whereas 1421 genes appeared down-regulated in the red-fleshed pummelo. A GO analysis assigned 11,983 differentially expressed genes to 53 subcategories, and 3582, 4722 and 3679 differentially expressed genes were involved in biological processes, cellular components and molecular functions, respectively. The mevalonate pathway involved in terpenoid backbone biosynthesis was significantly different between ‘Guanxi’ pummelo and its red-fleshed mutant, and the differences were closely associated with carotenoid biosynthesis. The expression levels of the carotenoid biosynthetic pathway genes were different between ‘Guanxi’ pummelo and its red-fleshed mutant, with the upstream genes having higher expression levels in red-fleshed pummelo than in ‘Guanxi’ pummelo, whereas the downstream gene β-CHX was downregulated. Two bHLH transcription factors (CL4778.Contig1 and CL4778.Contig2) were of the same branch as CubHLH1, regulating carotenoid biosynthesis in Citrus unshiu Marc., and Unigene CL4778.Contig2 had expression differences in two kinds of pummelo, which suggested CL4778.Contig2 should be a candidate gene controlling carotenoid accumulation in red pummelo. The study would be beneficial to identify the major gene regulating the red flesh trait.
Introduction
Pummelo (Citrus maxima) is an important species of the Citrus genus and a common germplasm for citrus breeding. It is widely commercially grown in some countries, including Japan, Thailand, Malaysia and China, partially because pummelo contains plentiful antioxidant compounds, such as carotenoids, flavonoids and limonoids [Citation1,Citation2]. In pummelo, the flesh colour of the fruit is a quality trait that correlates with nutritional value. Carotenoids are the major pigments responsible for the colour accumulation in Citrus fruit [Citation3–5]. Carotenoids impart the yellow, orange or red colouration that is characteristic of many fruit and required for optimum seed dispersal [Citation6,Citation7].
In higher plants, carotenoids are synthesized mainly in cell plastids [Citation8]. The carotenoid biosynthetic pathway plays various roles in photosynthesis, photoprotection, development, stress hormone responses, and the production of various volatiles and signalling apocarotenoids [Citation9]. The molecular mechanism of carotenoid biosynthesis is clear, and schematic diagrams of the main pathway steps have been built [Citation3,Citation4,Citation9,Citation10]. The first step involves the condensation of geranylgeranyl pyrophosphate, the C20 precursor, to build the colourless phytoene. This step is catalysed by a rate-limiting enzyme, phytoene synthase (PSY). Second, phytoene undergoes four consecutive desaturation reactions, catalysed by phytoene desaturase (PDS) and ζ-carotene desaturase (ZDS), and two isomerization reactions that are regulated by carotene isomerase (CRTISO) and 15-cis-ζ-carotene isomerase, to form lycopene. In addition, the cyclization of lycopene, as a key reaction, is mediated by two enzymes encoded by lycopene β-cyclase (β-LCY) and ε-LCY. Thus, two pathways exist, which result in the formation of β- and α-carotene. Lutein is formed from α-carotene catalysed by β-carotene hydroxylase (β-CHX) and ε-CHX. β-Carotene is first converted into zeaxanthin through a hydroxylation reaction catalysed by β-CHX, and then violaxanthin is generated through an epoxydation reaction catalysed by zeaxanthin epoxidase (ZEP). Finally, the end product of the pathway, abscisic acid, is generated from violaxanthin through a reaction mediated by 9-cis-epoxycarotenoid dioxygenase (NCED). Recently, the molecular bases of carotenoid biosynthesis and accumulation have been analysed. The accumulation of lycopene during fruit ripening results from the downregulation of β-LCY activity [Citation11]. Lycopene appears to be the determining factor in the red colouration of papaya flesh [Citation12]. Structural genes encoding enzymes of the carotenoid biosynthetic pathway, including PDS [Citation13–15], PSY [Citation14,Citation16,Citation17], ZDS [Citation14], β-LCY [Citation14], ε-LCY [Citation14, Citation17], ZEP [Citation14], CRTISO [Citation14] and HYb [Citation14], have been identified and characterized. A comprehensive available transcriptome dataset for pummelo has been obtained, and the associated molecular markers have been identified [Citation18]. Importantly, genes relating to secondary metabolites were mainly regulated by transcription factors [Citation19]. The carotenoid biosynthetic pathway is mainly regulated by the transcription factors MYB [Citation20,Citation21] and bHLH [Citation22–24].
Next-generation sequencing technology is a powerful and cost-effective tool for genome sequencing, genome re-sequencing, mRNA expression profiling and DNA methylation analyses. Recently, de novo transcriptome sequencing has been widely used for functional gene discovery in non-model organisms for which reference-genome information is lacking [Citation25]. In addition, this approach has also allowed the analyses of gene expression profiles using the quantification of short cDNA reads [Citation26–28]. In this manuscript, RNA sequencing (RNA-Seq) using next-generation sequencing was employed to determine the transcriptome differences in ‘Guanxi’ pummelo and its red-fleshed mutant to discover the key genes responsible for red flesh in pummelo.
Materials and methods
Plant materials
The materials used in this study were ‘Guanxi’ pummelo and its red-fleshed mutant, both of which are widely planted in Fujian Province, China. The fruit were collected at the ripe stage, with three biological replicates, and were immediately frozen in liquid nitrogen and kept at –80 °C until further use. Total RNA was extracted from the fruit employing the Tiangen RNAprep Pure Plant Kit (Tiangen, China) in accordance with the manufacturer’s protocol. Electrophoresis on a 1.0% (w/v) agarose gel demonstrated the quality of total RNA. The concentration of total RNA was determined by NanoDrop (Thermo Fisher Scientific, Wilmington, DE, USA).
Illumina sequencing
The cDNA libraries of the ‘Guanxi’ pummelo and red-fleshed pummelo fruit were prepared in line with the manufacturer’s instructions for mRNA-Seq sample preparation (Illumina Inc, San Diego, CA, USA). The cDNA library products were addressed by Illumina paired-end sequencing technology with read lengths of 150 bp, using an Illumina HiSeqXten-PE150 instrument (Novogene Bioinformatics Technology Co. Ltd., Beijing, China). The raw sequence reads were stored in the National Center for Biotechnology Information (NCBI) SRA database.
Sequence data filtering, de novo assembly and annotation
The raw data from the images were assembled with Illumina GA Pipeline 1.6 by removing low-quality reads (reads with unknown ‘N’ sequences, adaptor sequence fragments and empty reads). Next, the de novo assembly of the transcriptome into unique-sequence unigenes was processed with Trinity software, a short-read assembly program [Citation29]. In short, Trinity integrates reads with certain lengths of overlap to form longer fragments or contigs in the first place. Then, the reads are mapped back to contigs, and with paired-end reads, it becomes possible to detect contigs from the same transcript as well as the distances between these contigs. Finally, Trinity connects the contigs and generates sequences that can be extended on neither end. Such sequences are defined as unigenes [Citation29]. Later on, a Basic Local Alignment Search Tool (BLAST)x alignment (e-value < 0.00001) was carried out on the unigenes from protein databases, involving NCBI non-redundant protein (Nr) and nucleotide sequence (Nt), Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes (KEGG), Cluster of Orthologous Groups of proteins (COG), Interpro and Gene Ontology (GO). Also, the best alignments were employed to determine the sequence directions of the unigenes. If the results from the different databases showed a conflict between each other, then the priority order of Nr, Swiss-Prot, KEGG and COG was followed to determine the sequence direction. When a unigene was not aligned in any of the above databases, the software ESTScan was used to predict its coding regions and determine its sequence direction [Citation30]. In the final step, using Nr annotations, the Blast2GO program was used to obtain the GO and KEGG annotations for the unigenes [Citation31]. After the GO annotation was obtained for each unigene, WEGO software was used to classify the unigenes in terms of function and to determine the distribution of gene functions in the species at the macro level [Citation32]. A Venn diagram of the annotated proteins was constructed using the online software venny (http://bioinfogp.cnb.csic.es/tools/venny/index.html).
Analysis of differential gene expression
To map the differentially expressed reads, the sequenced raw data were filtered to remove low-quality reads. For read annotations, clean read sequences were mapped to our transcriptome reference database, allowing no more than a two-nucleotide mismatch [Citation29,Citation33]. Gene coverage is the percentage of a gene covered by reads. This value is equal to the ratio of the base number in a gene covered by unique mapping reads to the total base number of that gene. For the gene expression analysis, the number of expressed sequences was calculated and normalized to the number of Reads per Kb per Million reads.
To compare the differences in gene expression, the read frequency in each library was statistically analysed according to the method of Audic and Claverie [Citation33]. We used a false discovery rate < 0.001 and a |log2Ratio| > 1 as the threshold for judging the statistical significances of the gene expression differences [Citation34]. Next, the differentially expressed genes (DEGs) were subjected to GO and KEGG enrichment analyses [Citation35]. The genes related to carotenoid biosynthesis in KEGG pathways were collected and aligned to the unigenes from a transcriptome mixture of the fruit from ‘Guanxi’ pummelo and its red-fleshed mutant, using the BLASTx algorithm with an E-value of < 1 e − 5. The selected genes were: 1-deoxy-d-xylulose 5-phosphate synthase, 1-deoxy-d-xylulose 5-phosphate reductoisomerase, geranylgeranyl pyrophosphate synthase (GGPS), PSY, PDS, ZDS, CRTISO, β-LCY, ε-LCY, β-CHX, ZEP and NCED.
Bioinformatics analysis
The sequence alignments were conducted using the Vector NTI 10 software package (Thermo Fisher Scientific, Wilmington, DE, USA). The phylogenetic tree was constructed with MEGA5 software (Tokyo Metropolitan University, Hachioji, Tokyo, Japan) using the neighbour-joining method [Citation36].
Results and discussion
Sequencing and sequence assembly
RNA-Seq efficiently and conveniently enables rapid access to all transcripts from biological tissues. After filtering, 47,712,090 reads from the wild-type pummelo and 44,111,638 from the red-fleshed mutant remained, with a Q20 percentage of 96.69%. Trinity software was used to link the valid data to 46,425 unigenes, totalling 5,60,96,761 bp. The average length of each gene was 1208 bp (Supporting Information Table S1), which was longer than in another study [Citation18]. The N50 length was 1922 bp (Supporting Information Table S1). Accurate and effective assemblies usually have longer mean and N50 lengths [Citation37]. BLASTX software was employed to annotate the 46,425 assembled unigenes using the Nr, Nt, Swissprot, KEGG, COG, Interpro, GO and other protein databases (). A total of 40,261 proteins were obtained after removing repeated proteins. Of the predicted proteins, 68.40% had the highest homology to Citrus sinensis, 25.68% to Citrus clementina and 5.92% had closest genetic relationships to other species (Supporting Information Figure S1). Pummelo belongs to the Citrus genus, and the predicted proteins were highly homologous to its species.
Figure 1. Differentially expressed genes between ‘Guanxi’ pummelo and red-fleshed pummelo. The genes were divided into three classes. Red dots indicate genes that were upregulated, i.e. the gene’s expression in red-fleshed pummelo was higher than in ‘Guanxi’ pummelo. Blue dots indicate genes that were downregulated, i.e. the gene’s expression in ‘Guanxi’ pummelo was higher than in red-fleshed pummelo. Grey dots indicate genes that were not differentially expressed. The horizontal coordinates refer to the average FPKM value (A) and the vertical coordinates refer to the log2ratio (M).
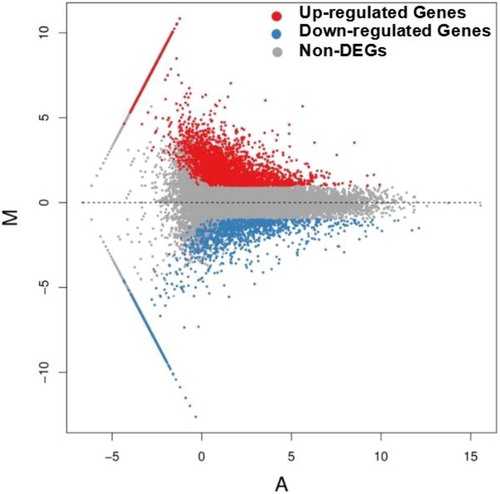
Table 1. Annotation summary of predicted protein numbers from various databases.
DEG analysis
To identify DEGs between ‘Guanxi’ pummelo and its red-fleshed mutant, putative DEGs were identified based on the FPKM values calculated from the read counts mapped onto the reference transcriptome. A total of 5143 unigenes were differentially expressed between ‘Guanxi’ pummelo and red-fleshed pummelo according to the comparison of expression levels with false discovery rate ≤ 0.001 and |log2Ratio| ≥ 1 (). The highest log2Ratio value reached 10.84, whereas the lowest value was –12.62. Using ‘Guanxi’ pummelo as a reference, 3722 upregulated unigenes (with higher levels of expression in the mutant red-fleshed pummelo) and 1421 downregulated unigenes (with higher levels of expression in ‘Guanxi’ pummelo) were identified. Significantly more unigenes were upregulated in its mutant than in ‘Guanxi’ pummelo.
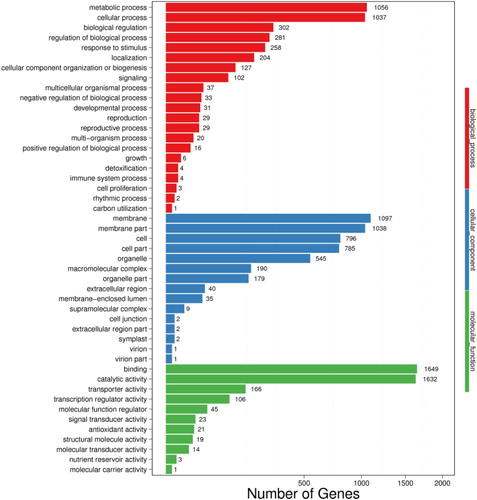
The GO analysis assigned 11,983 DEGs to 53 subcategories. Among them, 3582, 4722 and 3679 DEGs were involved in biological processes, cellular components and molecular functions, respectively. Within the biological process category, the majority were related to ‘metabolic processes’ (1056 DEGs) and ‘cellular processes’ (1037 DEGs). Within the cellular component category, most were enriched in the subcategories ‘membrane’ (1097 DEGs) and ‘membrane part’ (1038 DEGs). Within the molecular function category, the greater proportion was involved in ‘binding’ (1649 DEGs) and ‘catalytic activity’ (1632 DEGs; ). A total of 27,024 DEGs were annotated, and 4142 were mapped onto 130 pathways in the KEGG database. Most DEGs were involved in ‘metabolic pathways’ (2874), and ‘genetic information processing’ (692). The KEGG pathways ‘plant hormone signal transduction’, ‘ABC transporters’, ‘benzoxazinoid biosynthesis’, ‘diterpenoid biosynthesis’ and ‘carotenoid biosynthesis’ showed the greatest differences between ‘Guanxi’ pummelo and its red-fleshed mutant.
Figure 2. GO classifications of DEGs. Unigenes were assigned to three categories: cellular components, molecular functions and biological process.
The pathway ‘plant hormone signal transduction’ includes the signal transduction pathways of jasmonate, salicylate, auxin, cytokinin, gibberellin and abscisic acid. These plant hormones are believed to regulate fruit development and shape [Citation38–42]. The pathway ‘ABC transporters’ is mainly involved in plant defence and the symbiosis between plants and microorganisms [Citation43]. The pathway ‘benzoxazinoid biosynthesis’ plays an important role in host resistance against microbial pathogens and insects, and in allelopathic effects. The three pathways are not directly related to secondary metabolism. It is possible that they can indirectly regulate diterpenoid or carotenoid biosynthesis. Terpenoid backbone biosynthesis has two pathways, the mevalonate and the MEP/DOXP pathway. The mevalonate pathway showed significant differences between ‘Guanxi’ and red-fleshed pummelo, whereas no significant differences in the MEP/DOXP pathway. The mevalonate pathway produces two five-carbon building blocks, isopentenyl pyrophosphate and dimethylallyl pyrophosphate [Citation44], which are the basic building blocks of carotenoid biosynthesis [Citation45]. They undergo several reactions, which results in the production of the carotenoid precursor, geranylgeranyl pyrophosphate [Citation45]. Changes in the mevalonate pathway could activate the carotenoid biosynthesis pathway.
Unigenes in the carotenoid biosynthesis pathway and their expression pattern
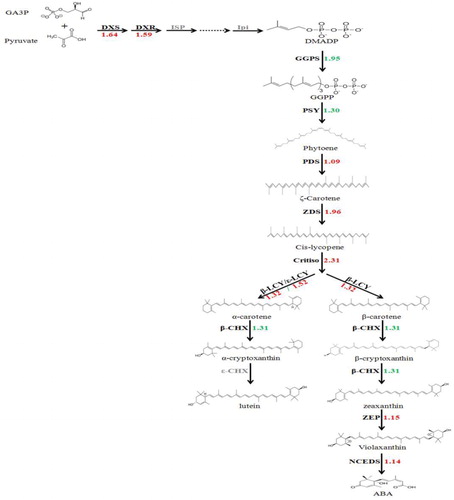
The FPKM values of genes were determined, and their relative expression levels were calculated in the carotenoid biosynthetic pathway. In total, 42 unigenes were involved in carotenoid biosynthesis. Although 12 genes related to carotenoid biosynthesis were used in the BLASTx analysis, only 1-deoxy-d-xylulose 5-phosphate synthase (DXS), 1-deoxy-d-xylulose 5-phosphate reductoisomerase (DXR), GGPS, PSY, PDS, ZDS, CRTISO, β-LCY, ε-LCY, β-CHX, ZEP and NCED have homologous genes in the assembled unigene database. The phylogenetic trees were constructed using the deduced amino acid sequences of 42 unigenes in pummelo and structural genes regulating the carotenoid biosynthesis in other species (Supporting Information Figures S2–S13). The same structural genes were assigned to the same branch (Figures S2–S13). The FPKM values of structural genes were determined to evaluate their relative expression levels. The carotenoid biosynthetic pathway was exhibited in pummelo, as described by Guo et al. [Citation22] ( and Supporting Information Table S2). The upstream structural genes had greater expression levels in red-fleshed pummelo than in ‘Guanxi’ pummelo, except GGPS and PSY, whereas the downstream structural gene β-CHX had lower expression levels in red-fleshed pummelo (). Only a few genes in other pathways were identified in this experiment. Genes homologous to ε-CHX were not identified in the unigene database.
Figure 3. Differences in the expression levels of genes in the carotenoid biosynthetic pathway.
Note: Arrows show the metabolic stream; left or upward arrows represent the genes catalysing the process; the light color indicates that these genes are in the assembled unigenes; the numbers represent the increased expression in red-fleshed pummelo compared with ‘Guanxi’ pummelo.
The key bHLH transcription factor regulating carotenoid biosynthesis
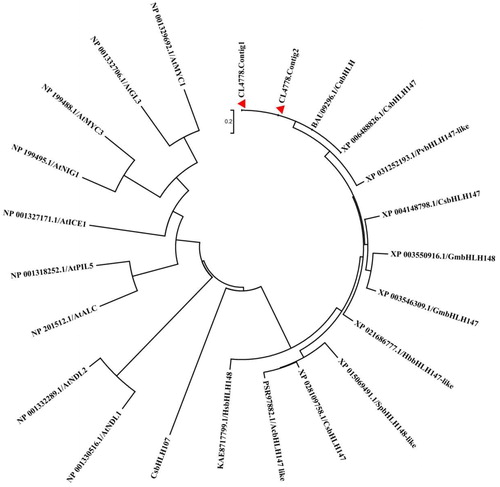
Previous studies demonstrated that transcription factors MYB and bHLH regulated carotenoid biosynthesis in Citrus species [Citation22,Citation23,Citation46,Citation47]. In this research, MYB protein CsMYB [Citation47] and bHLH protein CubHLH1 [Citation23] were used as the reference sequences for blastx analysis to identify the candidate genes regulating carotenoid biosynthesis. Five MYB transcript Unigenes were selected to construct the phylogenetic tree (). Phylogenetic analysis showed five MYB transcript Unigenes were not assigned to the same branch as CsMYB involved in carotenoid biosynthesis, suggesting that MYB could not be the key gene for the red flesh trait in the study (Supporting Information Figure S14). bHLH transcription factors negatively regulate carotenoid accumulation in Solanum lycopersicum [Citation23,Citation46]. CubHLH1 has a similar function to Arabidopsis AIFs and was directly involved in carotenoid metabolism in citrus fruit [Citation23]. Two bHLH transcriptor Unigenes (CL4778.Contig1 and CL4778.Contig2) were selected with scores over 1166 to construct the phylogenetic tree containing CsbHLH and the bHLH transcriptors from Arabidopsis and Citrus. CL4778.Contig1 and CL4778.Contig2 were assigned to the same branch as CubHLH1, which implies that the CmbHLH (CL4778.Contig2) in the carotenoid biosynthesis pathway had been isolated from the RNA-Seq database. In addition, the amino acid sequences of CL4778.Contig1 and CL4778.Contig2 were aligned with CubHLH1 (Supporting Information Figure S15). The deduced amino acid sequences alignment demonstrated that the typical bHLH domains of CL4778.Contig1 and CL4778.Contig2 were coincident with CubHLH1 [Citation18]. When compared with red flesh, CL1085.Contig2 had a higher transcript level in ‘Guanxi’ pummelo, as they were up-regulated 360 times. This suggested that the Unigene CL4778.Contig2 was the candidate gene for carotenoid biosynthesis, thus providing a theoretical basis for developing new cultivars with higher carotenoid levels.
Figure 4. Phylogenetic relationships of the deduced amino acid sequences of bHLH proteins in pummelo and bHLHs in other species.
Conclusions
In this study, transcriptome analysis was employed to discover the genetic differences between ‘Guanxi’ pummelo and its red-fleshed mutant. Here, a total of 46,425 unigenes, with an average length of 1208 bp, were assembled, in which 40,261 were annotated. A total of 5143 unigenes were differentially expressed between ‘Guanxi’ pummelo and red-fleshed pummelo, and 3722 upregulated unigenes (with higher levels of expression in the mutant red-fleshed pummelo) and 1421 downregulated unigenes (with higher levels of expression in ‘Guanxi’ pummelo) were identified. The mevalonate pathway involved in terpenoid backbone biosynthesis was significantly different, which was closely associated with carotenoid biosynthesis. The expression levels of the upstream genes had higher expression levels in red-fleshed pummelo than in ‘Guanxi’ pummelo, whereas the downstream gene β-CHX was downregulated in the carotenoid biosynthetic pathway. Two bHLH transcript factors, CL4778.Contig1 and CL4778.Contig2, were of the same branch as CubHLH1, regulating carotenoid biosynthesis, and Unigene CL4778.Contig2 had significant expression difference in the two kinds of pummelo. Therefore, we suggest that the bHLH transcript factor regulates the carotenoid accumulation in pummelo.
Supplemental Material
Download PDF (1.1 MB)Disclosure statement
The authors have declared that no competing interests exist.
Additional information
Funding
References
- Zhang M, Duan C, Zang Y, et al. The flavonoid composition of flavedo and juice from the pummelo cultivar (Citrus grandis (L.) Osbeck) and the grapefruit cultivar (Citrus paradisi) from China. Food Chem. 2011;129(4):1530–1536.
- Jayaprakasha GK, Girennavar B, Patil BS. Antioxidant capacity of pummelo and navel oranges: extraction efficiency of solvents in sequence. LWT-Food Sci Technol. 2008;41(3):376–384.
- Alquézar B, Zacarías L, Rodrigo MJ. Molecular and functional characterization of a novel chromoplast-specific lycopene beta-cyclase from Citrus and its relation to lycopene accumulation. J Exp Bot. 2009;60(6):1783–1797. [19325166]
- Guo F, Yu HW, Xu Q, et al. Transcriptomic analysis of differentially expressed genes in the Ras1CA-overexpressed and wildtype posterior silk glands. BMC Genom.. 2014;15:182.
- Ma G, Zhang LC, Witchulada YY, et al. Accumulation of carotenoids in a novel citrus cultivar 'Seinannohikari' during the fruit maturation. Plant Physiol Biochem. 2018;129:349–356.
- Bartley GE, Scolnik PA. Plant carotenoids: pigments for photoprotection, visual attraction, and human health. Plant Cell. 1995;7(7):1027–1038.
- Cuttriss AJ, Cazzonelli CI, Wurtzel ET, et al. Carotenoids. In: Rébeillé F, Douce R, editors. Biosynthesis of vitamins in plants. Amsterdam. The Netherlands: Elsevier; 2011. p. 1–36.
- Sun TH, Yuan H, Cao HB, et al. Carotenoid metabolism in plants: the role of plastids. Mol Plant. 2018;11(1):58–74.
- Shumskaya M, Wurtzel ET. The carotenoid biosynthetic pathway: thinking in all dimensions. Plant Sci. 2013;208(7):58–63.
- Lu SW, Zhang Y, Zhu KJ, et al. The citrus transcription factor CsMADS6 modulates carotenoid metabolism by directly regulating carotenogenic genes. Plant Physiol. 2018;176(4):2657–2676.
- Devitt LC, Fanning K, Dietzgen RG, et al. Isolation and functional characterization of a lycopene beta-cyclase gene that controls fruit colour of papaya (Carica papaya L.). J Exp Bot. 2010;61(1):33–39.
- Skelton RL, Yu Q, Srinivasan R, et al. Tissue differential expression of lycopene beta-cyclase gene in papaya. Cell Res. 2006;16(8):731–739.
- Kita M, Komatsu A, Omura M, et al. Cloning and expression of CitPDS1, a gene encoding phytoene desaturase in citrus. Biosci Biotechnol Biochem. 2001;65(6):1424–1428.
- Kato M, Ikoma Y, Matsumoto H, et al. Accumulation of carotenoids and expression of carotenoid biosynthetic genes during maturation in citrus fruit. Plant Physiol. 2004;134(2):824–837.
- Alquezar B, Rodrigo MJ, Zacarías L. Regulation of carotenoid biosynthesis during fruit maturation in the red-fleshed orange mutant Cara Cara. Phytochemistry. 2008;69(10):1997–2007.
- Tao N, Hu Z, Liu Q, et al. Expression of phytoene synthase gene (Psy) is enhanced during fruit ripening of Cara Cara navel orange (Citrus sinensis Osbeck). Plant Cell Rep. 2007;26(6):837–843.
- Li JW, Ma J, Feng K, et al. Carotenoid accumulation and distinct transcript profiling of structural genes involved in carotenoid biosynthesis in Celery. Plant Mol Biol Rep. 2018;36(4):663–674.
- Liang M, Yang X, Li H, et al. De Novo Transcriptome assembly of pummelo and molecular marker development. PLoS One. 2015;10(3):e0120615. [25799271]
- De Geyter N, Gholami A, Goormachtig S, et al. Transcriptional machineries in jasmonate-elicited plant secondary metabolism. Trends Plant Sci. 2012;17(6):349–359.
- Xu Q, Yu KQ, Zhu AD, et al. Comparative transcripts profiling reveals new insight into molecular processes regulating lycopene accumulation in a sweet orange (Citrus sinensis) red-flesh mutant. BMC Genom. 2009;10:540.
- Ampomah-Dwamena C, Thrimawithana AH, Dejnoprat S, et al. A kiwifruit (Actinidia deliciosa) R2R3-MYB transcription factor modulates chlorophyll and carotenoid accumulation. New Phytol. 2019;221(1):309–325.
- Guo F, Yu HW, Xu Q, et al. Transcriptomic analysis of differentially expressed genes in an orange-pericarp mutant and wild type in pummelo (Citrus grandis). BMC Plant Biol. 2015;15(1):44.
- Endo T, Fujii H, Sugiyama A, et al. Overexpression of a citrus basic helix-loop-helix transcription factor (CubHLH1), which is homologous to Arabidopsis activation-tagged bri1 suppressor 1 interacting factor genes, modulates carotenoid metabolism in transgenic tomato. Plant Sci. 2016;243:35–48.
- D'Amelia V, Raiola A, Carputo D, et al. A basic Helix-Loop-Helix (SlARANCIO), identified from a Solanum pennellii introgression line, affects carotenoid accumulation in tomato fruits. Sci Rep. 2019;9(1):3699.
- Liu D, Li S, Chen WJ, et al. Transcriptome analysis of purple pericarps in common wheat (Triticum aestivum L.). PLoS One. 2016;11(5):e0155428.
- Zhang N, Liu BL, Ma CY, et al. Transcriptome characterization and sequencing-based identification of drought-responsive genes in potato. Mol Biol Rep. 2014;41(1):505–517.
- Dorn KM, Fankhauser JD, Wyse D, et al. De novo assembly of the pennycress (Thlaspi arvense) transcriptome provides tools for the development of a winter cover crop and biodiesel feedstock. Plant J. 2013;75(6):1028–1038.
- Firon N, LaBonte D, Villordon A, et al. Transcriptional profiling of sweetpotato (Ipomoea batatas) roots indicates down-regulation of lignin biosynthesis and up-regulation of starch biosynthesis at an early stage of storage root formation. BMC Genom. 2013;14(1):460.
- Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–652.
- Iseli C, Jongeneel CV, Bucher P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol. 1999;99(2):138–148.
- Conesa A, Götz S, García-Gómez JM, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676.
- Mortazavi A, Williams BA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628.
- Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997;7(10):986–995.
- Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Statist. 2001;29(4):1165–1188.
- Kanehisa M, Goto S, Kawashima S, et al. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32(Database issue):D277–D280.
- Garg R, Patel RK, Tyagi AK, et al. De Novo assembly of Chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 2011;18(1):53–63.
- Tamura K, Peterson D, Peterson N, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–2739.
- Katsir L, Chung HS, Koo AJK, et al. Jasmonate signaling: a conserved mechanism of hormone sensing. Curr Opin Plant Biol. 2008;11(4):428–435.
- Johnson C, Boden E, Arias J. Salicylic acid and NPR1 induce the recruitment of trans-activating TGA factors to a defense gene promoter in Arabidopsis. Plant Cell. 2003;15(8):1846–1858.
- Kim T-W, Guan S, Sun Y, et al. Brassinosteroid signal transduction from cell-surface receptor kinases to nuclear transcription factors. Nat Cell Biol. 2009;11(10):1254–1260.
- Chapman EJ, Estelle M. Mechanism of auxin-regulated gene expression in plants. Annu Rev Genet. 2009;43:265–285.
- Mason MG, Mathews DE, Argyros DA, et al. Multiple type-B response regulators mediate cytokinin signal transduction in Arabidopsis. Plant Cell. 2005;17(11):3007–3018.
- Theodoulou FL. Plant ABC transporters. Biochim Biophys Acta. 2000;1465(1–2):79–103.
- Goldstein JL, Brown SB. Regulation of the mevalonate pathway. Nature. 1990;343(6257):425–430.
- Nisar N, Li L, Lu S, et al. Carotenoid metabolism in plants. Mol Plant. 2015;8(1):68–82.
- Zhu ZG, Chen GP, Guo XH, et al. Overexpression of SlPRE2, an atypical bHLH transcription factor, affects plant morphology and fruit pigment accumulation in tomato. Sci Rep. 2017;7(1):5786.
- Jiang CC, Zhang YF, Lin YJ, et al. Illumina® sequencing reveals candidate genes of carotenoid metabolism in three pummelo cultivars (Citrus maxima) with different pulp color. IJMS. 2019;20(9):2246.