
Abstract
Neural stem cell (NSC)-based gene therapies have been recently developed as effective strategies for treating brain tumors through inherent tumor tropism. However, this methodology has two considerable challenges: preventing NSCs from dying from the therapeutic agents encoded by their equipped genes before reaching tumor sites and the clearance of exogenous NSCs after therapeutic treatments. For these purposes, we established a novel doxycycline-inducible retroviral plasmid pTRE3G-TKGFP. A herpes simplex type 1 thymidine kinase (HSV1TK)-green fluorescent protein (GFP) fusion protein coding sequence was integrated into the multiple cloning site II (MCS II) of the pQcXIX vector, and the CMV IE promoter (PCMV IE) of pQcXIX was replaced with the doxycycline-inducible TRE3G promoter (PTRE3G). We then cloned the coding sequences of tumor necrosis factor-α (TNF-α), interleukin 6 (IL-6) and soluble interleukin 6 receptor (sIL-6R) into MCS I to combine HSV1TK (TK)/ganciclovir (GCV)-based suicide gene therapy against tumorigenic C6 glioma cells. TNF-α, IL-6 and sIL6R could efficiently induce tumorigenic C6 glioma cell differentiation, resulted in down-regulating the cell proliferation rate and the tumorigenicity of glioma cells and up-regulating the production of differentiation markers, such as connexin-43. Furthermore, gap junctions could enhance the bystander effect in suicide gene therapy. Consequently, we found that the retroviral plasmid transfected NSCs exerted stronger remedial effects on tumorigenic C6 glioma cells through the combination of differentiation and suicide gene therapy than by suicide gene therapy alone. This study is also the first case applying NSCs to conduct the combination of differentiation and TK/GCV-based suicide gene therapy on glioma cells.
Introduction
Brain cancers are lethal diseases in humans with poor prognosis. Among brain cancers, glioblastoma (GBM) is the most common and aggressive form. In America, approximately 10,000 new cases are diagnosed every year, and only 5.6% of patients survive 5 years post-diagnosis [Citation1]. Because the central nervous system (CNS) is relatively insulated, largely due to the blood-brain barrier (BBB), it is difficult to deliver therapeutic agents to the brain to cure tumors. Moreover, immune functionality is inhibited under this physiological condition, leading to immune evasion of tumor cells. Furthermore, GBM cells have the potential to infiltrate normal brain tissues. It is, therefore, difficult to cure GBM completely by conventional cancer remedies, including surgery, chemo- and radiation therapies [Citation2].
Clinically, the first case of gene therapy for a brain tumor was reported in 1992, which was conducted with a retrovirus equipped with the herpes simplex type 1 thymidine kinase gene (HSV1TK), produced in murine fibroblasts [Citation3]. Subsequently, manifold gene carriers, such as viruses, stem cells, cationic liposomes and nanoparticles, have been employed to develop therapeutic strategies. Stem cell-based gene therapies have reached the clinical stage [Citation4], and the tumor-tropic properties make stem cells superior to other vehicles for targeting tumors [Citation5]. Neural stem cells (NSCs), mesenchymal stem cells (MSCs), hematopoietic stem cells (HSCs), embryonic stem cells (ESCs), endothelial stem cells and induced pluripotent stem cells (iPSCs) can all home in on tumor sites through specific mechanisms [Citation6]. Moreover, stem cells can be engineered to constantly produce various therapeutic agents, thus overcoming difficulties in maintaining efficacious concentrations of anti-cancer drugs in malignant foci.
Multiple kinds of gene therapies have been developed to cure brain cancers, including suicide gene therapy, cytokine-mediated gene therapy, tumor suppressor gene therapy and oncolytic gene therapy [Citation7–10]. Among them, suicide gene therapy is the most frequently used approach to cure glioma both in preclinical studies and in clinical trials [Citation11]. The mechanism of this strategy involves converting a nontoxic prodrug into a cytotoxic agent by the specific suicide gene, which is usually not expressed or expressed at a low level in mammalian cells [Citation7]. Cytosine deaminase (CD)/5-fluorocytosine (5-FC) and HSV1TK (TK)/ganciclovir (GCV) are the most notable suicide gene/prodrug systems, transforming the tumor-localized 5-FC and GCV to active 5-fluorouracil (5-FU) and phosphorylated GCV, respectively; these phosphorylated compounds interfere with the synthesis of RNA and/or DNA to terminate the elongation of nucleic acid sequences, leading to cellular necrosis and/or apoptosis [Citation12,Citation13].
Stem cell-based gene therapy not only resolves the obstacles in tumor targeting but also enables stem cells to infiltrate the tumor mass for enhanced therapeutic effects [Citation5]. Nevertheless, this methodology also confronts several challenges. For example, stem cells must reach tumor sites before deterioration caused by therapeutic agents. Additionally, exogenous stem cells after treatments should be removed to prevent possible adverse effects on neighbouring normal cells because it has been validated that some multipotent and pluripotent stem cells, such as NSCs, MSCs, ESCs and iPSCs, can initiate or support malignancies under physiological conditions [Citation14].
To address these challenges, we established a doxycycline-inducible retroviral plasmid, pTRE3G-TKGFP, for stem cell-based combination gene therapy. This plasmid contains the doxycycline-inducible TRE3G promoter (PTRE3G) and a TK-green fluorescent protein (GFP) fusion protein coding sequence. Furthermore, previous studies have revealed that combination gene therapies exert additive or synergistic effects on brain tumors [Citation15,Citation16]. Accordingly, we have left an EcoRI cloning site of pTRE3G-TKGFP available for the insertion of other therapeutic genes to cooperate with the TK/GCV system in cancer therapy.
Under the hierarchical model of cancer stem cells (CSCs), they are a tumorigenic subpopulation of tumor cells and like normal stem cells, can self-renew and differentiate into heterogeneous cell lineages. As a result, they play critical roles in sustaining the malignancy of tumors by promoting tumor initiation, development and recurrence [Citation17]. It has been reported that differentiation therapy can induce the differentiation of CSCs to alleviate their tumorigenicity and make them more sensitive to common chemo- and radiation therapies. Currently, some successful cases have displayed the effects of differentiation therapy on CSCs [Citation18,Citation19]. For instance, bone morphogenetic protein 4 (BMP4) induces small mothers against decapentaplegic (Smad) signalling in human glioblastoma cells, which up-regulates the expression of neural differentiation markers, inhibits cellular proliferation and reduces the size of the tumorigenic CD133+ population. These findings have elucidated the inhibitory mechanisms by which BMP4 acts on brain CSCs [Citation18].
We intend to apply C17.2 NSCs against tumorigenic C6 glioma cells [Citation20] through a combination of differentiation and suicide gene therapy. The expression of therapeutic genes at specific time points can be manipulated by the doxycycline-inducible PTRE3G. Besides, the coding sequences of secreted TNF-α, IL-6 and sIL-6R, designed to perform differentiation therapy, were integrated into the EcoRI cloning site of pTRE3G-TKGFP. In previous studies, we confirmed these cytokines could efficiently induce differentiation in a tumorigenic C6 glioma cell line [unpublished observations]. This treatment resulted in down-regulating the cell proliferation rate and the tumorigenicity of glioma cells and up-regulating the production of differentiation markers, such as connexin-43, a major gap junction protein in the CNS [Citation21]. It has also been reported that gap junctions play a crucial role in the TK/GCV system-based suicide gene therapy, the functionality of which may be attributed to the enhancement of the bystander effect [Citation22,Citation23]. The more gap junctions established between cells in a tumor mass, the greater number of cells eradicated by the administration of GCV. Because of these observations, we anticipate a better therapeutic effect exerted by the combination of differentiation and suicide gene therapy on tumorigenic C6 glioma cells. In this study, we chose C17.2 NSCs to carry multiple therapeutic genes for combination gene therapy; these cells can home in on glioma cells in vitro and in vivo [Citation24,Citation25].
Materials and methods
Cell cultures and restriction enzymes
The rat C6 glioma cell line was obtained from the National Health Research Institute Cell Bank (Zhunan, Taiwan). The tumorigenicity of this C6 rat glioma cell line has been previously evaluated [Citation20]. These cells were cultured in Dulbecco's Modified Eagle Medium (DMEM)/F12 supplemented with 10% (w/v) fetal bovine serum (FBS) and 1% (w/v) penicillin and streptomycin (Invitrogen, Waltham, MA, USA) in a humidified atmosphere with 5% CO2 at 37 °C. Besides, C17.2 NSCs (Sigma-Aldrich, St. Louis, MO, USA) and GP2-293 packaging cells (Clontech, Mountain View, CA, USA) were maintained in DMEM supplemented with 10% FBS, and 1% penicillin and streptomycin in a humidified atmosphere with 5% CO2 at 37 °C. All restriction enzymes were purchased from Promega (Madison, WI, USA) except for PacI (New England Biolabs, Hitchin, UK).
Western blot analysis
After being treated with cytokine complexes, including IL-6, a combination of IL-6 and soluble IL-6 receptor (IL-6/sIL-6R) and TNF-α plus IL-6/sIL-6R (TNF-α/IL-6/sIL-6R) (TNF-α: 20 ng/mL, IL-6: 5 ng/mL and sIL-6R: 100 ng/mL; all from PeproTech, Rocky Hill, NJ, USA), for 2 days, C6 glioma cells were lysed using CelLytic M (Sigma-Aldrich) and centrifuged at 15,000 g for 15 min at 4 °C. Protein concentrations were measured by the Bradford method (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. Afterwards, 20 μg of proteins from different experimental groups were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes (Bio-Rad) followed by blocking with 5% (w/v) non-fat milk for 1 h at room temperature. The membranes were then incubated with specific primary antibodies against connexin-43 (Cell Signaling Technology, Danvers, MA, USA) and β-actin (Merck Millipore, Darmstadt, Germany) overnight at 4 °C. After washing with Tris buffered saline (TBS) with 0.1% (w/v) Tween-20, the membranes were treated with appropriate horseradish peroxidase-labelled secondary antibodies (Santa Cruz Biotechnology, Dallas, TX, USA), and then the Western blots were incubated with enhanced chemiluminescence (ECL) detection reagents (Thermo Fisher Scientific, Waltham, MA, USA). Finally, the bands were visualized using a UVP BioSpectrum image system (UVP, Upland, CA, USA).
Construction of doxycycline-inducible retroviral plasmid
Plasmid construction was divided into three steps: insertion of TK-GFP, doxycycline-inducible PTRE3G and the therapeutic genes, including TNF-α, IL-6 and sIL-6R, for differentiation therapy. The coding sequence of TK from pORF-HSV1tk (InvivoGen, San Diego, CA, USA) without its stop codon, was first amplified with a high fidelity polymerase, Herculase II Fusion DNA Polymerase (Agilent Technologies, Santa Clara, CA, USA), by polymerase chain reaction (PCR) using a T100 thermal cycler (Bio-Rad). The primer sequences were as follows: 5′-GAGAATTCATGGCCTCGT-3′ (forward) and 5′-GAGTCGACCAGTTAGCCT-3′ (reverse). The PCR procedure included an initial denaturation step (94 °C for 5 min), 30 cycles of amplification (94 °C for 30 s, 50 °C for 30 s and 72 °C for 45 s) and an extension step (72 °C for 10 min), consecutively. This sequence was then cut with EcoRI and SalI and ligated into EcoRI/SalI-digested pAcGFP1-N1 (Clontech) with T4 DNA ligase (Promega) according to the manufacturer’s instructions. DNA sequencing was used to determine the sequence of pHSV1tk-AcGFP.
The complementary DNA (cDNA) of the TK-GFP fusion protein was then cloned into the multiple cloning site II (MCS II) of pQcXIX (Clontech). The primers for amplifying the coding sequence of TK-GFP were designed as follows: 5′-ATGATAAGCTTGATCAATGGCCTCGTACCCCGGC-3′ (forward) and 5′-CCACTGATATCTCGAGGCTCACTTGTACAGCTCAT-3′ (reverse). The PCR procedure included an initial denaturation step (94 °C for 5 min), 30 cycles of amplification (94 °C for 30 s, 65 °C for 30 s and 72 °C for 70 s) and an extension step (72 °C for 10 min), consecutively. The TK-GFP open reading frame (ORF) was flanked by BclI and XhoI restriction sites and then ligated with BclI/XhoI-digested pQcXIX by an In-Fusion HD Cloning Kit (Clontech) according to the manufacturer’s instructions. DNA sequencing was used to determine the sequence of pQcXIX-TKGFP.
The CMV IE promoter (PCMV IE) of pQcXIX was subsequently replaced by the doxycycline-inducible PTRE3G. The pQcXIX-TKGFP was linearized with XbaI and PacI, and then the In-Fusion HD Cloning kit was used to combine it with PTRE3G amplified with forward primer: 5′-GGATCGATCCTCTAGACTCGAGTTTACTCCCT-3′ and reverse primer: 5′-ATTCCGGATCCGTTAATTAACGACTTTACGAG-3′. The PCR procedure included an initial denaturation step (94 °C for 5 min), 30 cycles of amplification (94 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s) and an extension step (72 °C for 10 min), consecutively. DNA sequencing was used to determine the sequence of pTRE3G-TKGFP.
The coding sequences of TNF-α, IL-6 and sIL-6R for differentiation therapy were finally integrated into the EcoRI cloning site of pTRE3G-TKGFP. These cDNAs were respectively obtained from pUNO1-hTNFA, pUNO1-hIL06 and pUNO1-hIL06Ra (all from InvivoGen). To prepare secreted cytokines, the N-terminal cDNA of TNF-α was replaced with a commercial leader sequence, Ig-κ chain: 5′-ATGGAGACAGACACACTCCTGCTATGGGTACTGCTGCTCTGGGTTCCAGGTTCCACTGGTGAC-3′; the cDNA of sIL-6R was then obtained by deleting the C-terminal sequence of IL-6Rα. The stop codon in the cDNA sequences of TNF-α and sIL-6R were removed. Besides, 2 A peptide sequences were introduced to connect three cDNA sequences, in the order sIL6R-P2A-TNFα-T2A-IL6 [Citation26]. These modifications were carried out by PCR. The primer sequences were as follows: 5′-AACGGATCCGGAATTCTGAGATCACCGGTA-3′ (sIL-6R forward), 5′-CAGCCTGCTTCAGCAGGCTGAAGTTAGTAGCTCCGCTTCCCAGTGGTACTGAA-3′ (sIL-6R reverse); 5′-GTACTGCTGCTCTGGGTTCCAGGTTCCACTGGTGACGTCAGATCATC-3′ (TNF-α forward 1), 5′-AAGGACACCATGGAGACAGACACACTCCTGCTATGGGTACTGCTGCT-3′ (TNF-α forward 2), 5′-AAGCAGGCTGGAGACGTGGAGGAGAACCCTGGACCTAAGGACACCATGGA-3′ (TNF-α forward 3), 5′-ACCGCATGTTAGCAGACTTCCTCTGCCCTCTCCGCTTCCCAGGGCAATG-3′ (TNF-α reverse); 5′-CTGCTAACATGCGGTGACGTCGAGGAGAATCCTGGACCTATGAACTCCTTC-3′ (IL-6 forward), 5′-GAGAGGGGCGGAATTCACAATCTGAGGTGCCCATGCTACATTTGCCGAAGA-3′ (IL-6 reverse). TNF-α cDNA sequence was first amplified with TNF-α forward 1/TNF-α reverse and further elongated with TNF-α forward 2/TNF-α reverse and TNF-α forward 3/TNF-α reverse in sequence. The PCR procedure included an initial denaturation step (94 °C for 5 min), 30 cycles of amplification (94 °C for 30 s, 68 °C for 30 s and 72 °C for 60 s) and an extension step (72 °C for 10 min), consecutively. Finally, these modified cDNA sequences were simultaneously ligated into EcoRI-digested pTRE3G-TKGFP by an In-Fusion HD Cloning kit. DNA sequencing was used to determine the sequence of pTRE3G-sIL6R-TNFα-IL6-IRES-TKGFP.
Retroviral system
The Retro-X system (Clontech) was employed to produce retroviruses in the GP2-293 packaging cell line. One day before transfection, GP2-293 cells were seeded to 10 cm plates at a density of 5 × 106 cells/plate in 10 mL of growth medium. After cells were cotransfected with 15 µg of retroviral plasmid (pQcXIX-TKGFP or pRetroX-TetOne-Puro) and 15 µg of envelope plasmid (pVSV-G) by Xfect Transfection Reagent (Clontech) for 4 h, the transfection medium was replaced with fresh complete growth medium for further 48 h of cell incubation. The retroviral supernatants were harvested, filtered through a 0.45-μm filter (Sigma-Aldrich) to remove cellular debris, condensed with Retro-X Concentrator (Clontech) reagent according to the manufacturer’s instructions and finally resuspended in DMEM/F12 or DMEM as stock solutions. Subsequently, the viral titre was determined with a QuickTiter Retrovirus Quantitation Kit (Cell Biolabs, San Diego, CA, USA) according to the manufacturer’s instructions. To obtain a suitable infectious dose, tumorigenic C6 glioma cells and C17.2 NSCs were respectively infected with serial dilutions of specific retroviral stock solutions for 24 h to find the maximal concentration that did not affect the viability of the transduced cells.
Cytotoxicity assay
After retrovirus (produced from the pQcXIX-TKGFP-transfected GP2-293 cells) infection, tumorigenic C6 glioma cells were added to 6-well plates at a density of 2 × 105 cells per well and cultured in the medium containing GCV (10 µg/mL; InvivoGen). Two days later, the cell population of each group was inspected under light microscopy (40× field; Olympus, Tokyo, Japan) and determined by 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assays (Merck Millipore). The medium in each well was replaced with 500 µL MTT solution (5 mg/mL), and the cells were incubated for an additional 4 h. Subsequently, the solution was discarded, and formazan blue was dissolved in 2 mL dimethyl sulfoxide (DMSO; Sigma-Aldrich). The optical density was measured at 540 nm.
Cell sorting
After retroviral transduction for 2 days, tumorigenic C6 glioma cells were trypsinized, washed and resuspended in PBS (106 cells/mL). Cell suspensions were filtered through 40-μm Falcon strainers (BD Biosciences, San Jose, CA, USA). Subsequently, tumorigenic C6 glioma cells were analysed with FlowJo Software (V.10.0.8r1; FlowJo LLC, Ashland, OR, USA), and the high GFP-expressing cells (top 1.8%) were isolated by a FACSCalibur flow cytometry system (BD Biosciences). Afterwards, these cells were cultured for 24 h at 37 °C and then observed with fluorescence microscopy (200× field; Olympus, Tokyo, Japan).
Functional analysis of PTRE3G
C17.2 NSCs were seeded in 24 well dishes at a density of 4 × 104 cells/well in 250 µL of growth medium. One day later, cells were transfected with pCMV-tet3G and/or pTRE3G-TKGFP (total 1 μg of plasmid DNA) by Xfect Transfection Reagent and cultured in the medium with or without doxycycline (1 μg/mL) for 48 h. Finally, the fluorescence expressed by cells was examined by fluorescence microscopy (40× field).
Puromycin selection
Twenty-four hours after retrovirus (produced from the pRetroX-TetOne-Puro-transfected GP2-293 cells) infection, C17.2 NSCs were cultured in the medium containing puromycin (10 µg/mL; InvivoGen), which was replaced every 3 days. Drug-resistant colonies appeared two weeks later, and they were transferred to 6-well plates (one colony per well) for an additional one-week incubation. Reverse transcription PCR (RT-PCR) was applied to examine the expression of the Tet-On 3 G transactivator in each colony. Total RNA of the treated cultures was isolated using the Total RNA Extraction Miniprep System (Viogene, New Taipei City, Taiwan) according to the manufacturer’s instructions and quantified by measuring the absorbance at 260 nm (Nanodrop 1000; Thermo Fisher Scientific). Five micrograms of total RNA from different experimental groups were then reverse transcribed using the SuperScript III First-Strand Synthesis System (Invitrogen) following the manufacturer’s instructions. One microliter (250 ng) of newly synthesized cDNA from each group was amplified in a 50 μL reaction mixture comprising 5 μL of 10× Taq buffer (Protech, Taipei, Taiwan), 1 μL of 10 mmol/L dNTPs (Protech), 1 μL of 100 μmol/L forward primer (MDBio, Taipei, Taiwan), 1 μL of 100 μmol/L reverse primer (MDBio) and 0.5 μL of Pro Taq DNA polymerase (Protech) using a T100 thermal cycler. Two primers were employed as follows: 5′-CAGGCATCATACCCACTCCT-3′ (forward) and 5′-GTCAGCAGGCAGCATATCAA-3′ (reverse). The PCR procedure included an initial denaturation step (94 °C for 5 min), 30 cycles of amplification (94 °C for 30 s, 55 °C for 30 s and 72 °C for 30 s) and an extension step (72 °C for 10 min), consecutively.
Combination of differentiation and suicide gene therapy
Tumorigenic C6 glioma cells were treated with combined differentiation and suicide gene therapy with C17.2 (Tet-On 3 G) cells. C17.2 (Tet-On 3 G) cells were first seeded in 6-well plates at a density of 1.5 × 105 cells per well and incubated for 24 h. Subsequently, C17.2 NSCs were transfected with 6 μg of pTRE3G-TKGFP or 6 μg of pTRE3G-sIL6R-TNFα-IL6-IRES-TKGFP by Xfect Transfection Reagent for 4 h and then incubated with glioma cells (2.5 × 105 cells per well) in DMEM supplemented with tetracycline-free FBS (Clontech) and doxycycline (1 µg/mL; Clontech). Twenty-four hours later, prodrug GCV (10 µg/mL) was added into the medium, and the cells were maintained for another 24 h. MTT assays were then employed to evaluate the total cell population of each group. The medium in each well was replaced with 500 µL MTT solution (5 mg/mL), and the cells were incubated for an additional 4 h. Subsequently, the solution was discarded, and formazan blue was dissolved in 2 mL DMSO. The optical density was measured at 540 nm.
Statistical analysis
Statistical analysis was performed using IBM SPSS Statistics version 26 (IBM Corporation, Armonk, NY, USA). All data are presented as the mean values with standard deviation (± SD) of at least three independent experiments or are representative of experiments repeated at least three times. The statistical significance of the results from the cytotoxicity assay was analysed by paired Student’s t-test. In addition, one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was employed to evaluate differences among distinct experimental groups in combination of differentiation and suicide gene therapy. Differences were considered statistically significant at a level of p < 0.05.
Results and discussion
Upregulation of connexin-43 in tumorigenic C6 glioma cells by TNF-α/IL-6/sIL-6R
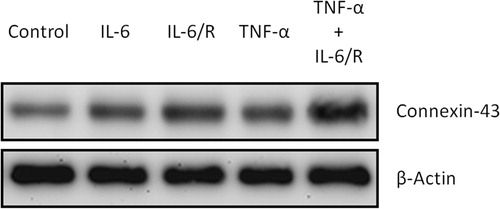
C6 glioma cells were treated with different cytokine complexes, including IL-6, IL-6/sIL-6R and TNF-α/IL-6/sIL-6R, for 2 days, and the level of connexin-43 was then evaluated by Western blotting assay. The blots showed that TNF-α/IL-6/sIL-6R could more efficiently upregulate connexin-43 production than IL-6 alone and IL-6/sIL-6R (). Therefore, the TNF-α/IL-6/sIL-6R complex was applied as the candidate for differentiation therapy of combination gene therapy. Additionally, IL-6 alone and IL-6/sIL-6R respectively elicited minor and moderate effects on connexin-43 in glioma cells.
Figure 1. Upregulation of connexin-43 in tumorigenic C6 glioma cells by IL-6, IL-6/sIL-6R and TNF-α/IL-6/sIL-6R. Western blots of glioma cells were carried out after cytokine treatment.
Construction and functional analysis of pQcXIX-TKGFP
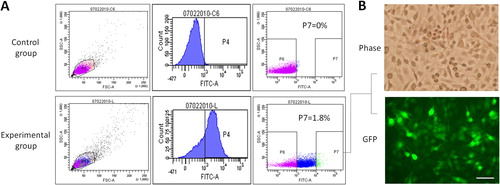
Herein, we selected TK/GCV as a suicide gene/prodrug system and fused TK with GFP. GFP not only supplies a selection marker for constructing the retroviral plasmid but also labels the retrovirus-transduced cells for fluorescence imaging. The retroviral plasmid pQcXIX is the backbone of pTRE3G-TKGFP. First, the cDNA of TK was amplified from pORF-HSV1tk without its stop codon and integrated into the MCS (EcoRI/SalI) of pAcGFP1-N1 (; Supplemental data Citation1: pHSV1tk-AcGFP vector sequence). The coding sequences of TK and GFP are in the same reading frame. TK-GFP cDNA from pHSV1tk-AcGFP was then amplified and cloned into the MCS II (BclI/XhoI) of pQcXIX (; Supplemental data Citation2: pQcXIX-TKGFP vector sequence). To verify the functionality of the TK-GFP fusion protein and to establish a retroviral packaging system, pQcXIX-TKGFP was employed to manufacture retroviruses. After determination of the titre of the retrovirus, tumorigenic C6 glioma cells were transduced with various amounts of retrovirus to determine the favourable infectious dose, which turned out to be ≤4 × 1010 viral particles per millilitre (VP/mL) (). The serial dilutions of the retroviral stock solution were used to infect glioma cells, and they were then incubated in the medium containing GCV for 48 h. Finally, MTT assays were applied to evaluate the total cell population of each group. The results revealed that the more TK-GFP cells produced, the fewer glioma cells survived (), demonstrating the functionality of TK in the TK-GFP fusion protein. Subsequently, tumorigenic C6 glioma cells transduced by retroviruses (4 × 1010 VP/mL) with high expression of GFP were enriched using a cell sorter () and then examined under fluorescence microscopy (). The observed fluorescence proved the functionality of GFP in the TK-GFP fusion protein.
Figure 2. Construction of TK-GFP fusion protein. (A) Construction of the coding sequence of TK without a stop codon, derived from pORF-HSV1tk, into the MCS of pAcGFP1-N1. (B) Integration of the TK-GFP ORF, obtained from pHSV1tk-AcGFP, into the MCS II of pQcXIX.
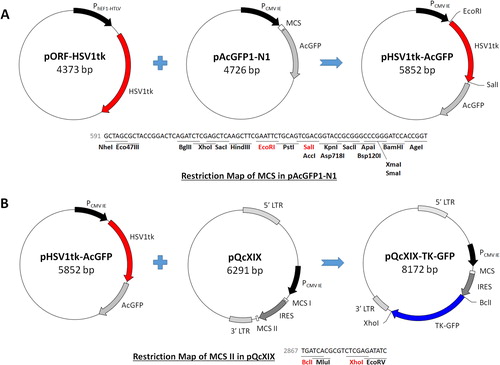
Figure 3. Functional analysis of TK in TK-GFP fusion protein. (A) Determination of the retroviral infectious dose. Plasmid pQcXIX-TKGFP was employed to produce retroviruses. A range of retrovirus concentrations to infect tumorigenic C6 glioma cells was used to determine a suitable infectious dose of ≤4 × 1010 VP/mL. (B) Functional analysis of TK. Glioma cells were infected with serial dilutions of the retroviral stock solution and incubated in the medium containing GCV (10 μg/mL) for 48 h. The cell populations were quantified by MTT assays. (C) Phase-field images; scale bar, 500 μm. The statistical results are the mean of three experiments ± SD, and the asterisk indicates significant differences (p < 0.05) between retrovirus-transduced and control groups.
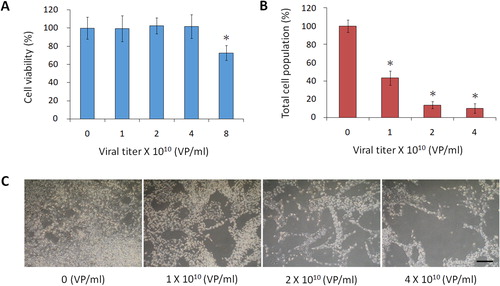
Figure 4. Functional analysis of GFP in TK-GFP fusion protein. (A) Isolation of GFP-expressing cells. None (control group) and 4 × 1010 VP/mL retrovirus-infected cells (experimental group) were sorted by flow cytometry to enrich for those highly expressing GFP (gate P7). (B) Inspection of the fluorescence in glioma cells. Isolated glioma cells were incubated for 24 h and then observed with fluorescence microscopy; scale bar, 80 μm.
Construction and functional analysis of pTRE3G-TKGFP
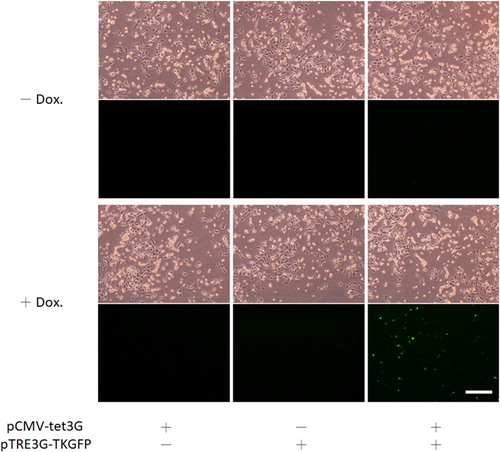
To control the expression of therapeutic genes at the desired time point, the doxycycline-inducible promoter PTRE3G, derived from pTRE3G-IRES, was used to replace the PCMV IE (XbaI/PacI) in pQcXIX-TKGFP (; Supplemental data Citation3: pTRE3G-TKGFP vector sequence). Doxycycline, a synthetic tetracycline derivative, in a complex with the Tet-On 3 G transactivator, can drive PTRE3G to express downstream genes. Upon binding of these two factors, the conformation of the Tet-On 3 G transactivator was changed, allowing it to bind to the tet operator sequences of PTRE3G. After construction, the functionality of PTRE3G was further examined. C17.2 NSCs were transfected with pCMV-tet3G and/or pTRE3G-TKGFP and incubated in media with or without doxycycline for 48 h. Finally, the expression of GFP in the transfected cells was examined by fluorescence microscopy (). Fluorescence was observed only when the C17.2 NSCs contained both pCMV-tet3G and pTRE3G-TKGFP and were cultured with doxycycline because the expression of GFP was driven by PTRE3G. These results validated the functionality of the promoter in pTRE3G-TKGFP. Therefore, the doxycycline-inducible retroviral plasmid for stem cell-based combination gene therapy was ready.
Figure 5. Construction of pTRE3G-TKGFP. The PCMV IE sequence of pQcXIX-TKGFP was replaced by the PTRE3G sequence derived from pTRE3G-IRES.
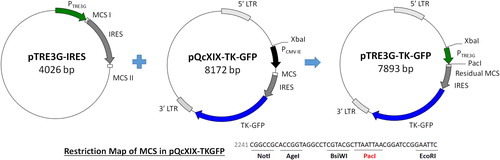
Figure 6. Functional analysis of PTRE3G in pTRE3G-TKGFP. C17.2 NSCs were transfected by pCMV-tet3G and/or pTRE3G-TKGFP and incubated in the media with or without doxycycline (1 μg/mL) for 48 h. The fluorescence expressed by pCMV-tet3G and pTRE3G-TKGFP cotransfected cells was inspected under microscopy; scale bar, 250 μm. Dox. represents doxycycline.
Construction of pTRE3G-sIL6R-TNFα-IL6-IRES-TKGFP
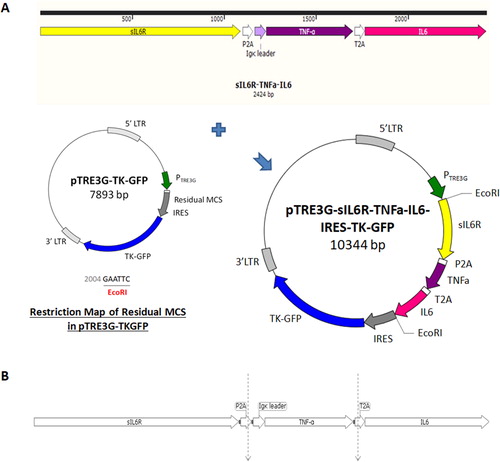
Genes for differentiation therapy were integrated into pTRE3G-TKGFP. TNF-α and sIL-6R were manipulated into secreted cytokines by modifying their cDNA through the PCR-based technique. The 228-base pair (bp) sequence encoding the N-terminus of TNF-α was replaced by an Ig-κ chain leader sequence; the cDNA sequence encoding the C-terminus of IL-6Rα was then removed, yielding sIL-6R. Subsequently, three cDNAs, including TNF-α, IL-6 and sIL-6R, were each elongated with 2 A peptide sequences and simultaneously inserted into the EcoRI site of residual MCS of pTRE3G-TKGFP (; Supplemental data Citation4: pTRE3G-sIL6R-TNFα-IL6-IRES-TKGFP vector sequence). Therefore, functional PTRE3G could initiate the expression of TNF-α, IL-6 and sIL-6R in the microenvironments containing Tet-On 3 G transactivator and doxycycline. Because the stop codon of TNF-α and sIL-6R were removed, the construct contains a long ORF. During translation, the polypeptide chain is interrupted by the 2 A peptide sequences, leading to the cleavage between glycine and the last proline at its C-terminus () [Citation27].
Figure 7. Construction of three cytokine genes for differentiation therapy into pTRE3G-TKGFP. (A) Preparation of pTRE3G-sIL6R-TNFα-IL6-IRES-TKGFP. The cDNAs of TNF-α, IL-6 and sIL-6R were separately elongated with 2 A peptide sequences and then simultaneously integrated into the EcoRI site of Residual MCS of pTRE3G-TKGFP in the order sIL6R-P2A-TNFα-T2A-IL6. (B) Formation of 3 individual peptide fragments during polypeptide chain synthesis due to the 2 A peptide sequences.
Tumorigenic C6 glioma cells treated with combined differentiation and suicide gene therapy by C17.2 NSCs
A stable C17.2 cell line, C17.2 (Tet-On 3 G), was prepared to constantly express Tet-On 3 G transactivator. GP2-293 cells were transduced with pRetroX-TetOne-Puro to produce retroviruses. After determination of the retroviral titre, C17.2 NSCs were infected with serial dilutions of the retroviral stock solution to find a suitable infectious dose. The maximal concentration was chosen without exerting cytotoxic effects. The retrovirus-transduced C17.2 NSCs were subsequently screened by puromycin to select cells expressing the Tet-On 3 G transactivator. RT-PCR was finally performed to check the expression of Tet-On 3 G transactivator in these cells ().
Figure 8. Cytotoxicity effect exerted by C17.2 NSCs through differentiation therapy combined with suicide gene therapy. (A) Stable cell line C17.2 (Tet-On 3 G) examined with RT-PCR to confirm the expression of Tet-On 3 G transactivator. The size of the amplified fragment is 526 bp. (B) The effect of combination gene therapy on C6 glioma cells. C17.2 (Tet-On 3 G) cells were transfected with pTRE3G-TKGFP or pTRE3G-sIL6R-TNFα-IL6-IRES-TKGFP and cocultured with tumorigenic C6 glioma cells in the medium containing doxycycline (1 μg/mL). Following 24 h incubation, GCV (10 µg/mL) was selectively added to the culture medium. The total cell populations were finally evaluated with MTT assays. The statistical results are the mean of three experiments ± SD, and the asterisk indicates significant differences (p < 0.05) between groups.
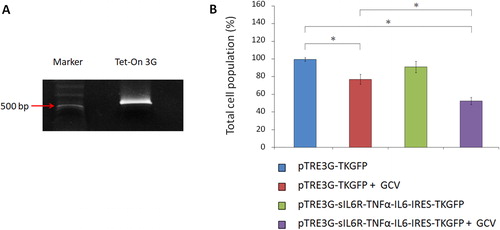
C17.2 (Tet-On 3 G) NSCs were finally employed to perform the suicide gene therapy or the combination of differentiation and suicide gene therapy on tumorigenic C6 glioma cells. After C17.2 (Tet-On 3 G) NSCs were transfected with pTRE3G-TKGFP or pTRE3G-sIL6R-TNFα-IL6-IRES-TKGFP for 4 h, these cells were incubated with glioma cells in the medium containing doxycycline. After 24 h, the prodrug GCV was added into the culture media. Following another 24 h of incubation, the cell populations were measured with MTT assays. According to the results, the total cell population both in the suicide gene therapy group (pTRE3G-TKGFP + GCV) and in the combination gene therapy group (TRE3G-sIL6R-TNFα-IL6-IRES-TKGFP + GCV) showed significant differences (p < 0.05) with the control group (). Besides, the combination gene therapy reduced the cell viability (47.2 ± 4.2%; mean ± SD, n = 3) more efficiently than the suicide gene therapy alone (22.6 ± 5.6%; mean ± SD, n = 3) as expected.
Future potential
A doxycycline-inducible retroviral plasmid, pTRE3G-TKGFP, was successfully constructed for stem cell-based combination gene therapy. This plasmid consists of the doxycycline-inducible PTRE3G, EcoRI cloning site, IRES and the coding sequence for a TK-GFP fusion protein. The expression of PTRE3G-driven genes could be initiated through administration of doxycycline to the Tet-On 3 G transactivator localized microenvironments. Other therapeutic genes could be integrated into the unoccupied EcoRI cloning site for combination therapy with the TK/GCV system. Furthermore, IRES supplies another translational initiation site. As a result, the expression of TK-GFP will not be disturbed by the insertion of other genes into the EcoRI cloning site. GFP contributes significantly to retroviral plasmid construction as a selection marker and could be used in an optical imaging system to label stem cells. For TK, it phosphorylates the prodrug GCV, converting it into a cytotoxic substance that interferes with DNA elongation, leading to cellular apoptosis. TK could also be applied for positron emission tomography (PET) imaging as a kinase to activate radiolabelled probes. This real-time, noninvasive monitoring technique makes it possible to determine the location and assess the survival of TK-expressing stem cells in physiological environments, allowing us to determine the appropriate time to administer GCV for suicide gene therapy. Moreover, PET shows better properties than optical imaging techniques in terms of tissue penetration and imaging resolution [Citation28]. Taken together, pTRE3G-TKGFP represents a powerful and convenient tool for future research on a doxycycline-inducible stem cell-based combination gene therapy. Using this plasmid for therapeutic strategies simply involves incorporating genes of interest into the EcoRI cloning site of pTRE3G-TKGFP.
Herein, we combined differentiation therapy and the TK/GCV-based suicide gene therapy systems to treat tumorigenic C6 glioma cells. It has been demonstrated that upon differentiation, the production of gap junction protein is up-regulated. Gap junctions allow the exchange of small cytosolic molecules (<1 kDa) between adjacent cells [Citation29]. Because the molecular masses of mono-, di- and triphosphorylated GCV are all below 550 Da, phosphorylated GCV could be transferred from TK-expressing stem cells to neighbouring cells through gap junctions [Citation30]. Consequently, the formation of these intercellular channels increases the efficacy of TK/GCV-based suicide gene therapy. According to the results in this study, a better cytotoxic effect was shown by the combination of differentiation and suicide gene therapy than by suicide gene therapy alone.
It is difficult to precisely delineate the differences between CSCs and normal stem cells because of their manifold similarities, and normal stem cells can even initiate tumors in vivo due to their innate self-renewal and pluripotency [Citation14,Citation31]. As a result, the clearance of exogenous stem cells after therapy is essential to prevent side effects affecting adjacent normal cells. In our combination gene therapy system, the suicide gene TK is expressed by the stem cells. Upon phosphorylation of GCV by TK, triphosphorylated GCV competes with endogenous deoxyguanosine triphosphate (dGTP) in transcription, resulting in apoptosis. Finally, the exogenous stem cells are eliminated after treatment, resolving safety concerns in combination gene therapy in physiological environments.
It has been suggested recently that tumors may contain multiple subclones, and each has its CSCs with unique phenotypic or genetic features [Citation17,Citation32]. As a result, the ultimate goal of cancer therapy is to eliminate the CSCs of each subclone in tumor masses. Unfortunately, therapies designed to target specific CSC markers may not be effective because they cannot eliminate all malignant CSCs. Nonetheless, differentiation therapy, in which the tumorigenicity of CSCs is reduced by inducing differentiation, is still a viable option for eliminating CSCs. Therefore, the combination of differentiation and suicide gene therapy may be a suitable alternative for better efficacy in curing CSCs.
Stem cells have intrinsic pathotropic properties and thus could selectively deliver therapeutic agents to cancer cells. Furthermore, they could be engineered to express and/or release long-lasting therapeutic agents at the site of malignancy. These advantages resolve the main limitations of conventional chemo- and radiation therapies, which inevitably cause a considerable loss of healthy cells and have unfavourable pharmacokinetic properties. Stem cell-based combination gene therapy is a promising tool for future cancer therapy and might be further improved by modification of stem cell properties, development of novel combination strategies and increasing the resolution and tissue penetration of real-time noninvasive imaging systems.
Conclusions
In this study, a novel doxycycline-inducible retroviral plasmid, pTRE3G-TKGFP, was established for NSC-based combination gene therapy to conquer two considerable challenges: preventing NSCs dying from the therapeutic agents encoded by their equipped genes before reaching tumor sites and the clearance of exogenous NSCs after therapeutic treatments. Besides, the coding sequences of secreted TNF-α, IL-6 and sIL-6R, designed to perform differentiation therapy, were integrated into the EcoRI site of residual MCS of pTRE3G-TKGFP to combine TK/GCV-based suicide gene therapy against tumorigenic C6 glioma cells. Our findings verify NSC-based combination of differentiation and TK/GCV-based suicide gene therapy is a potential methodology for an In vitro tumorigenic C6 glioma model.
Supplemental Material
Download PDF (670.3 KB)Supplemental Material
Download PDF (353.6 KB)Supplemental Material
Download PDF (349.1 KB)Supplemental Material
Download PDF (690.9 KB)Acknowledgements
Inn-Ray Chu carried out his thesis research under the auspices of the Graduate Program of Biotechnology in Medicine, National Tsing Hua University and National Health Research Institutes. We also thank Mr. Chao-Yang Hsiao (Cell Sorter Core Facility of National Health Research Institutes) for his technical support in flow cytometry applications, and Mr. Li-Kun Huang (National Tsing Hua University) for the diagnosis on all figures.
Disclosure statement
The authors declare that they have no competing interests.
Availability of data and materials
The data used during the current study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Ostrom QT, Gittleman H, Truitt G, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the united states in 2011-2015. Neuro-Oncology. 2018;20(suppl_4):iv1–iv86.
- Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol. 2006;1:97–117.
- Culver KW, Ram Z, Wallbridge S, et al. In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors. Science. 1992;256(5063):1550–1552.
- Portnow J, Synold TW, Badie B, et al. Neural stem cell-based anticancer gene therapy: a first-in-human study in recurrent high-grade glioma patients. Clin Cancer Res. 2017;23(12):2951–2960.
- Aboody KS, Brown A, Rainov NG, et al. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc Natl Acad Sci USA. 2000;97(23):12846–12851.
- Stuckey DW, Shah K. Stem cell-based therapies for cancer treatment: separating hope from hype. Nat Rev Cancer. 2014;14(10):683–691.
- Duarte S, Carle G, Faneca H, et al. Suicide gene therapy in cancer: where do we stand now? Cancer Lett. 2012;324(2):160–170.
- Colombo MP, Trinchieri G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002;13(2):155–168.
- Kim SS, Harford JB, Moghe M, et al. A tumor-targeting nanomedicine carrying the p53 gene crosses the blood-brain barrier and enhances anti-PD-1 immunotherapy in mouse models of glioblastoma. Int J Cancer. 2019;145(9):2535–2546.
- Geletneky K, Hajda J, Angelova AL, et al. Oncolytic H-1 parvovirus shows safety and signs of immunogenic activity in a first phase I/IIa glioblastoma trial. Mol Ther. 2017;25(12):2620–2634.
- Mooney R, Hammad M, Batalla‐Covello J, et al. Concise review: neural stem Cell-Mediated Targeted Cancer Therapies. Stem Cells Transl Med. 2018;7(10):740–747.
- Mullen CA, Kilstrup M, Blaese RM. Transfer of the bacterial gene for cytosine deaminase to mammalian cells confers lethal sensitivity to 5-fluorocytosine: a negative selection system. Proc Natl Acad Sci USA. 1992;89(1):33–37.
- Beltinger C, Fulda S, Kammertoens T, et al. Herpes simplex virus thymidine kinase/ganciclovir-induced apoptosis involves ligand-independent death receptor aggregation and activation of caspases. Proc Natl Acad Sci USA. 1999;96(15):8699–8704.
- Lee AS, Tang C, Rao MS, et al. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat Med. 2013;19(8):998–1004.
- Park J, Kim CG, Shim JK, et al. Effect of combined anti-PD-1 and temozolomide therapy in glioblastoma. Oncoimmunology. 2019;8(1):e1525243.
- Cammarata FP, Torrisi F, Forte GI, et al. Proton therapy and Src family kinase inhibitor combined treatments on U87 human glioblastoma multiforme cell line. IJMS. 2019;20(19):4745.
- Polyak K, Hahn WC. Roots and stems: stem cells in cancer. Nat Med. 2006;12(3):296–300.
- Piccirillo SG, Reynolds BA, Zanetti N, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444(7120):761–765.
- Basile MS, Mazzon E, Krajnovic T, et al. Anticancer and differentiation properties of the nitric oxide derivative of lopinavir in human glioblastoma cells. Molecules. 2018;23(10):2463. Sep 26
- Fang KM, Yang CS, Lin TC, et al. Induced interleukin-33 expression enhances the tumorigenic activity of rat glioma cells. Neuro-oncology. 2014;16(4):552–566.
- Nakase T, Naus CC. Gap junctions and neurological disorders of the central nervous system. Biochim Biophys Acta. 2004;1662(1-2):149–158.
- Asklund T, Appelskog IB, Ammerpohl O, et al. Gap junction-mediated bystander effect in primary cultures of human malignant gliomas with recombinant expression of the HSVtk gene. Exp Cell Res. 2003;284(2):185–195.
- Li S, Tokuyama T, Yamamoto J, et al. Bystander effect-mediated gene therapy of gliomas using genetically engineered neural stem cells. Cancer Gene Ther. 2005;12(7):600–607.
- Schichor C, Birnbaum T, Etminan N, et al. Vascular endothelial growth factor A contributes to glioma-induced migration of human marrow stromal cells (hMSC). Exp Neurol. 2006;199(2):301–310.
- Jurvansuu J, Zhao Y, Leung DS, et al. Transmembrane protein 18 enhances the tropism of neural stem cells for glioma cells. Cancer Res. 2008;68(12):4614–4622.
- Shaimardanova AA, Kitaeva KV, Abdrakhmanova II, et al. Production and application of multicistronic constructs for various human disease therapies. Pharmaceutics. 2019;11(11):580. Nov 6
- Sharma P, Yan F, Doronina VA, et al. 2A peptides provide distinct solutions to driving stop-carry on translational recoding. Nucleic Acids Res. 2012;40(7):3143–3151.
- Yaghoubi SS, Campbell DO, Radu CG, et al. Positron emission tomography reporter genes and reporter probes: gene and cell therapy applications. Theranostics. 2012;2(4):374–391.
- Kumar NM, Gilula NB. The gap junction communication channel. Cell. 1996;84(3):381–388.
- Spray DC, Hanstein R, Lopez-Quintero SV, et al. Gap junctions and bystander effects: good Samaritans and executioners. Wiley Interdiscip Rev Membr Transp Signal. 2013;2(1):1–15.
- Papaccio F, Paino F, Regad T, et al. Concise review: cancer cells, cancer stem cells, and mesenchymal stem cells: influence in cancer development. Stem Cells Transl Med. 2017;6(12):2115–2125.
- Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275–291.