
Abstract
As a well-known edible fungus that generally grows at low temperature, Flammulina velutipes is believed to be able to withstand most undesirable microbes. However, in this study, we discovered and isolated cold-adapted bacteria that developed rapidly and caused soft rot in the stipe of F. velutipes. The pathogen was identified as Mycetocola saprophilus based on morphology, 16S rRNA sequences, and multilocus sequence typing analysis. To further understand the pathogenic mechanisms of soft rot disease in F. velutipes, we sequenced and analyzed the genome of strain JXN-3 using PacBio sequencing. The de-novo assembly of the genome of strain JXN-3 was ∼3.47 Mb, with a GC content of 66.32%. The genome was composed of 3276 genes, of which 3094 were protein coding. Many of the identified protein-coding genes were associated with the synthesis of cell wall-degrading enzymes and antifungal compounds, including chitin deacetylase, chitinase, 1,4-beta-D-glucan glucohydrolase, beta-glucanase, endoglucanase Acf2, exo-beta-1,3-glucanase and bacteriocin. Compared with other members of Mycetocola, we also found a JXN-3-unique gene cluster for synthesizing bacteriocin. These genes may contribute to the pathogenesis of soft rot in F. velutipes stipes. Future studies should further investigate the involvement of these proteins in the incidence and progression of soft rot disease in F. velutipes.
Introduction
Flammulina velutipes (Curt. ex Fr.) Sing. is a familiar edible fungus, commonly known as the Enoki mushroom, Lily mushroom, Futu mushroom, or golden needle mushroom [Citation1]. This fungus mainly grows in East Asia, including China, Japan, Korea and Vietnam. China is the largest producer of F. velutipes worldwide, with an annual production of 2.4 million tons [Citation2]. White F. velutipes, which has a long white stalk and a small cap, is the main cultivated variety of this species and is popular in the mushroom market [Citation3].
Bacterial diseases are the main causes of significant losses in commercial cultivation, as such pathogens tend to accumulate in the production environment [Citation4]. The bacterial diseases of cultivated mushroom include yellow blotch, brown blotch, internal stipe necrosis, black rot and soft rot disease [Citation5–10]. Several bacteria are known to cause soft rot disease in cultivated mushrooms. For example, Burkholderia gladioli pv. agaricicola infection leads to soft rot disease in several cultivated mushroom, including Agaricus bisporu, Pleurotus ostreatus, F. velutipes [Citation11,Citation12]; Erwinia carotovora subsp. carotovora infection also leads to soft rot disease in F. velutipes [Citation13]. In addition, infection with Pantoea sp. results in severe soft rot disease in Pleurotus eryngii [Citation14]. During the early stages of infection, typical symptoms of soft rot disease include water-soaked lesions on the fruiting bodies. Ultimately, diseased fruiting bodies stop growing and soft rot develops, accompanied by an offensive odor [Citation11–14].
F. velutipes is a low-temperature edible fungus; 20–24 °C is suitable for F. velutipes mycelia, and 10–15 °C is optimal for fruit body formation and development [Citation15]. However, soft rot disease caused by a cold-adapted bacterium potentially threatens the Chinese F. velutipes cultivation industry [Citation4].
To further understand the pathogenic mechanisms of soft rot disease, the complete genomes of some pathogenic bacteria strains have recently been sequenced and analyzed, including that of Erwinia persicina B64, which causes soft rot in onions [Citation16], and that of Dickeya dianthicola ME23, which causes soft rot in potatoes [Citation17]. However, the pathogenic mechanisms of soft rot disease in F. velutipes have not been elucidated.
In this study, we isolated and identified the soft rot pathogen in F. velutipes, using morphological observations, 16S ribosomal RNA sequence data and multilocus sequence typing analysis. We then sequenced and analyzed the genome of the causal agent of soft rot disease using PacBio sequencing. We also identified several protein-coding genes associated with the synthesis of cell wall-degrading enzymes and antifungal compounds.
Materials and methods
Isolation of the causal bacteria
F. velutipes samples with typical bacterial rot symptoms were collected from three mushroom farms in Hebei Province, China. The symptoms and prevalence of the bacterial diseases were investigated, observed and recorded at the three sampled farms. Several pieces of infected mushroom stipes (about 0.5 g in total) were surface sterilized by soaking in 75% ethanol for 30 s. Stipe pieces were then washed three times with sterile water and suspended in 4.5 mL of sterile distilled water [Citation18]. The suspensions were serially diluted, plated onto nutrient agar (NA) medium and incubated at 25 °C for 48 h. Morphologically distinct colonies were purified by streaking on the same medium. The isolation frequencies of stipe pieces exhibiting early symptoms of infection were determined. To initially quantify the pathogenicity of each isolate, isolates were tested against the mycelium of F. velutipes.
Infectivity test
Pathogenicity tests in F. velutipes were conducted to determine whether the pathogen fulfilled the stipulations of Koch’s Postulates. In brief, bacterial isolates from the diseased samples were cultured separately in NA at 25 °C in a shaker at 150 rpm for 48 h, and concentrations were adjusted to 1 × 108 cfu/mL for use as the inoculums in the pathogenicity tests. In total, four different bacterial isolates were obtained.
The F. velutipes used in this experiment were obtained from a local mushroom house in Hebei Province, China. All selected mushrooms were homogenous in size and healthy. The first infection trial was performed by spraying separate groups of healthy F. velutipes with one of each of the bacterial suspensions, using sterile water as the control. After inoculation, the mushrooms were incubated for 5 d at 20 °C and examined for symptoms. Isolates that caused soft rot symptoms in the first trial were reassayed in a second infection trial, following the same procedure. Bacteria causing soft rot in the second infection trial were re-isolated from symptomatic F. velutipes by serial dilutions as described above. The strain identical to the original inoculum was considered a causal agent of soft rot.
Morphological characteristics
We grew the identified pathogenic bacterial strain on potato dextrose agar (PDA), King’s medium B (KMB), King’s medium A (KMA) and brain-heart infusion broth (BHI) at 25 °C for 48 h to observe its morphological characteristics. After 5 d, the bacterium cultured on KMB medium was irradiated under an ultraviolet (UV) lamp at a wavelength of 300 nm to observe the production of fluorochrome. A pure colony of this pathogenic bacterium was transferred to a 100-mL flask containing 20 mL of liquid BHI medium and placed on a rotary shaker at 150 rpm for 24 h for Gram staining. The size and morphology of the bacterium were examined using a Hitachi S-4800 scanning electron microscope (SEM, Hitachi, Tokyo, Japan). The bacterium was preliminarily identified based on morphological characteristics and Gram staining.
The pathogenic bacterium was then inoculated into a 100-mL flask containing 20 mL of liquid BHI medium and placed on a rotary shaker at 150 rpm for 24 h. We then transferred the culture suspension to liquid BHI medium to a final concentration of 1%. We then recorded the growth of the pathogenic strain over 24 h under a variety of conditions: altering the pH of the growth medium with hydrochloric acid (pH 4, 5, 6, 7, 8, 9, 10, 11, 12 or 13); varying the incubation temperature (15 °C, 20 °C, 25 °C, 30 °C or 35 °C); or changing the NaCl concentration in the growth medium (0%, 1%, 2%, 3%, 4% or 5%). This experiment was performed three times.
16s rRNA gene sequencing and multilocus sequence analysis
Genomic DNA was extracted from the pathogenic bacterium using the Bacterial Genomic DNA Kit EE161 (TransGen Biotech, Beijing, China). The 16S rRNA gene was amplified using polymerase chain reaction (PCR) with the universal primers 27F (5′-AGAGTTTGATCCTGGCTCA-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′) [Citation19]. PCRs were performed using the Taq DNA Polymerase Kit AP141 (TransGen Biotech, Beijing, China), with the following cycling conditions: 94 °C denaturation for 3 min; followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 30 s, extension at 72 °C for 2 min; and a final extension at 72 °C for 10 min. The amplified product was electrophoresed on 1% (w/v) agarose gels, purified using a PCR Purification Kit (TransGen Biotech, Beijing, China), and cloned with a T3 Cloning Kit (Beijing TransGen Biotech Co., Ltd). Recombinant plasmids were obtained from overnight LB cultures using the Plasmid Miniprep Kit (TransGen Biotech, Beijing, China), and were sent to Beijing Genomics Institute (BGI, Beijing, China) for sequencing.
The housekeeping genes coding for DNA gyrase subunit B (gyrB), RNA-polymerase subunit B (rpoB), recombinase A (recA), and polyphosphate kinase (ppk) were also PCR amplified using the primers listed in as previously described [Citation20,Citation21].
Table 1. Primers used for the amplification and sequencing of the housekeeping genes.
The JXN-3 16S rRNA, gyrB, rpoB recA and ppk gene sequences (accession nos. MG837010, MH029125, MH029127, MH023303 and MH029126, respectively) were searched against GenBank with BLAST [Citation22] to identify similar taxa. We then downloaded 16S rRNA, gyrB, rpoB recA and ppk sequences of representative bacterial isolates of the family Microbacteriaceae from GenBank (), and aligned the sequences of each gene using MAFFT v7.427 [Citation23]. Aligned genes were concatenated for each organism to increase the phylogenetic information. A phylogeny based on the concatenated alignment was constructed using the maximum-likelihood (ML) method in FastTree [Citation24]. One-thousand bootstrap replications were conducted [Citation25].
DNA preparation and genome sequencing
The pathogenic strain was cultivated at 25 °C in BHI for 24 h, and then genomic DNA was extracted using the Genomic DNA Mini Kit (Qiagen, Hilden, Germany). The concentration and quality of the DNA were measured using a Nanodrop (Thermo Fisher Scientific, Waltham, MA, USA) and 1% agarose electrophoresis, respectively. The total amount of DNA was greater than 5 μg, and the concentration was greater than 100 ng/μL. A 10-kb library was constructed using a SMRTbell Template Prep kit (Pacific Biosciences, CA, USA), following the manufacturer’s instructions. Following quality control of the library using an Agilent 2100 Bioanalyzer and a Qubit 4.0 fluorometer (Invitrogen, Carlsbad, CA, USA), the library was sequenced using the PacBio RS II platform with P6-C4 chemistry (Pacific Biosciences, CA, USA).
Genome assembly and annotation
The assembly of the genome of the pathogenic strain was performed using the PacBio SMRT Analysis 2.3.0 HGAP 2 software [Citation26], and merged using Minimus2 [Citation27]. The Arrow algorithm in SMRT Analysis 2.3.0 was used to make single-base corrections [Citation26]. Protein-coding genes (CDS) were predicted using GlimmerHMM v3.02 [Citation28]. tRNAscan-SE v1.3.1 [Citation29] was used to identify transfer RNAs (tRNAs), RNAmmer v1.2 [Citation30] was used to identify rRNAs, and Rfam 12.0 [Citation31] was used to identify non-coding RNAs (ncRNAs). Repeat sequences were identified using RepeatMasker v4.0.9 [Citation32], and tandem repeats (TRs) were identified using Tandem Repeats Finder v4.07b [Citation33]. MinCED [Citation34] was used to detect clustered regularly interspaced short palindromic repeat sequences (CRISPRs), and simple sequence repeats (SSRs) were predicted using MISA v1.0 [Citation35]. Genomic base modifications and motif analyses were performed using the pbsmrtpipe.pipelines.ds_modification_motif_analysis module of the SMRT Link software [Citation36].
Based on preliminary findings, which indicated that the pathogenic strain fell into the Mycetocola, the reference genomes of M. miduiensis CGMCC1.11101 (NZ_FOVM00000000) [Citation37], M. reblochoni REB411 (NZ_FUKR00000000) [Citation38], M. saprophilus 32,480 M. saprophilus NRRLB-24119 (NZ_JOEC01000000) [Citation39], M. tolaasinivorans IF016277 (NZ_RCUX01000000) [Citation40], M. lacteus JCM11654 (NZ_RCUY01000000) [Citation40], M. reblochoni JCM30549 (NZ_FUKR00000000) [Citation38], Mycetocola sp. 449 (NZ_CP026949), Mycetocola sp. 622 (NZ_QEFB01000000), M. zhadangensis ZD14 (NZ_RCWJ00000000) [Citation41] and M. manganoxydans CCTCCAB209002 (NZ_RCUV00000000) [Citation42] were downloaded from GenBank.
Custom Python and Perl scripts were used to calculate the average nucleotide identity (ANI) of each gene. Pan-genome analyses of the isolated pathogenic strains, M. lacteus, M. manganoxydans, M. miduiensis, M. reblochoni, M. saprophilus and M. tolaasinivoran, were performed with Roary [Citation43].
Genome annotation was performed against the Clusters of Orthologous Groups (COG) database [Citation44]. Comprehensive analyses of genomic COG annotations for pathogenic bacteria and other members of the genus Mycetocola were performed. Carbohydrate-active enzymes (CAZymes) in the pathogenic strain and in other members of Mycetocola were identified with the dbCAN2 meta server, using a Hidden Markov Model (HMM) (http://bcb.unl.edu/dbCAN2/) [Citation45].
Secondary metabolites
Gene clusters associated with the biosynthesis of secondary metabolites were identified using AntiSMASH (version 5.0, https://antismash.secondarymetabolites.org/#!/about) [Citation46], with a cutoff e-value of 1 × 10−5. To determine their genomic synteny, we compared JXN-3 with M. saprophilus 32,480 M. saprophilus NRRLB-24,119 M. reblochoni JCM30549, M. reblochoni REB411 and M. tolaasinivorans IF016277. Multiple sequence alignment of the secondary metabolite gene cluster was performed by MAFFT v7.427 [Citation23]. The phylogenetic tree was used for ML analysis in IQ-TREE [Citation47] with 1000 bootstrap replicates.
Results and discussion
Isolation of the soft rot pathogen from F. velutipes
Winter mushrooms exhibiting soft rot symptoms were observed at a Chinese mushroom farm during 2015 and 2017. Small brown spots were first observed on stipes at a distance of 25–45 mm from the cap (about 1/3 of the length of the stipe), followed by the development of water-soaked lesions on the stipe, and eventually leading to soft rot accompanied by an offensive odor (). Four morphologically distinct bacterial colonies of various relative sizes were isolated from the tissues of F. velutipes exhibiting with early symptoms of soft rot. Of these four bacteria, the strain JXN-3 was the most effective inhibitor of F. velutipes mycelia, with a separation frequency of 86% ().
Figure 1. Isolation of the soft rot pathogen from Flammulina velutipes. (a) Symptoms of soft rot disease; (b) the inhibition of F. velutipes mycelial growth by the isolated bacterial strain JXN-3; (c) F. velutipes treated with strain JXN-3; (d) re-isolation of JXN-3 from inoculated F. velutipes; (e) strain JXN-3 cultured on potato dextrose agar (PDA); (f) strain JXN-3 cultured on King’s medium A (KMA); (g) strain JXN-3 cultured on King’s medium B (KMB); (h) strain JXN-3 cultured on brain heart infusion (BHI); (i) gram-staining of strain JXN-3; and (j) scanning electron micrograph of strain JXN-3. Scale bars: 500 nm.
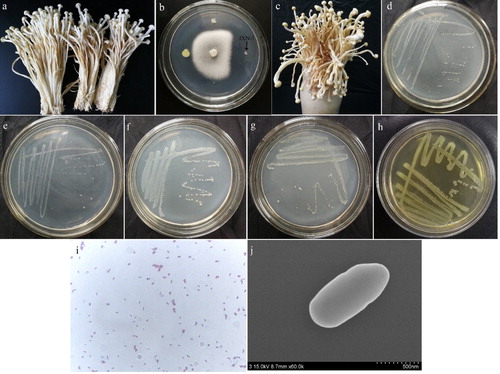
In the first pathogenicity assay, F. velutipes inoculated with strain JXN-3 exhibited typical symptoms of soft rot (). No symptoms of soft rot were observed in F. velutipes inoculated with the other three bacterial isolates or in the control group. Thus, we performed a second infection trial with strain JXN-3. Consistent with the first trial, all F. velutipes inoculated with strain JXN-3 during the second trial presented with identical soft rot symptoms. The bacterium re-isolated from the diseased F. velutipes () was identical to the original inoculum (JXN-3). This satisfied Koch’s Postulates, and indicated that bacterial strain JXN-3 caused soft rot in F. velutipes.
Taxonomic identification of JXN-3
The pure colonies of strain JXN-3 grown on PDA () and KMA () were milky-white and opaque, had smooth surfaces with circular margins, and grew well under aerobic conditions. Those grown on KMB () were yellowish-white, and no pigment production was observed under UV light after 5 d. Conversely, the colonies cultured on BHI () were yellow. Gram staining of strain JXN-3 was positive (). Under SEM, cells of strain JXN-3 were non-motile, rod-shaped and non-sporulating; cells were 4.6–6 μm wide and 6–14 μm long ().
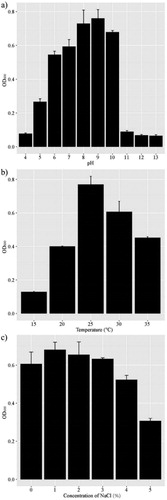
Growth analyses indicated that strain JXN-3 grew in media with a pH range of 4–13, with an optimal pH of 9 (), and at temperatures of 15–35 °C with an optimum of 25 °C (). Salinity tolerance tests showed that the optimal NaCl concentration for JXN-3 growth was 1%; at a NaCl concentration of ≥5%, the growth rate decreased noticeably (). Strain JXN-3 grew at low temperatures (15 °C), similar to other species of Mycetocola [Citation37,Citation38,Citation41,Citation42] and to F. velutipes, probably because these organisms inhabit similar environments.
Figure 2. Effects of pH (a), temperature (b) and salinity (c) on the growth of strain JXN-3. Bars represent the mean ± standard deviation of three different experiments.
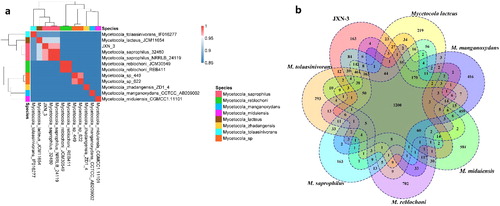
The 16S rRNA sequence of strain JXN-3 was 1361 bp long and had 99.79% similarity to the 16S rRNA sequence of M. saprophilus DSM 15178 (GenBank accession no. NR_024678.1; [Citation40]; ). The housekeeping genes of strain JXN-3 (gyrB, rpoB, recA and ppk) were 558 bp, 1045 bp, 654 bp and 1051 bp, respectively, and were 97.52%, 98.95%, 96.79% and 95.72% similar to the gyrB, rpoB, recA and ppk genes of M. saprophilus DSM 15178 (). The phylogenetic tree recovered strain JXN-3 in a well-supported monophyletic clade with M. saprophilus (). Therefore, we preliminarily identified JXN-3 as M. saprophilus based on morphological, 16S rRNA and MLSA analyses.
Figure 3. Maximum likelihood phylogeny of strain JXN-3 and other bacterial strains based on concatenated 16S rRNA, gyrase subunit B (gyrB), RNA-polymerase subunit B (rpoB), recombinase A (recA), and polyphosphate kinase (ppk) gene sequences, with heat map showing sequence similarity to strain JXN-3. Numbers at nodes represent bootstrap values based on 1000 replicates. The scale bar represents the number of substitutions per site.

Properties of the genome of strain JXN-3
We produced 118,598 subreads containing 810,141,733 bp bases. The de-novo assembly of strain JXN-3 yielded a genome of 3,468,784 bp, with a GC content of 66.32%. The genomic sequence of JXN-3 has been submitted to GenBank (accession No. CP041635). In the JXN-3 genome, we predicted 3276 genes, of which 3094 were CDS. The JXN-3 genome encoded 13 rRNAs, 56 tRNAs and 113 ncRNA (). In addition, we predicted 11 TRs, 9 SSRs and 23 CRISPRs, representing, in total, 1.51% of the entire genome. Furthermore, 12,778 modified bases were detected in the genome, including 4535 N6-methyl-adenine bases, 7918 N4-methyl-cytosine bases and 12,778 bases with no specific epigenetic modification (). The genome of strain JXN-3 consists of one circular 3,468,784 bp chromosome (see graphical genome map in , including COG categories and modified bases).
Figure 4. Circular genome map of the single chromosome of strain JXN-3, and color-coded list of Clusters of Orthologous Groups (COG) categories. Rings from inner to outer: %GC content, with GC content higher than average in green, and GC content lower than average in dark blue; GC skew [(G − C)/(G + C)], with positive in red and negative in purple; ncRNAs (tRNA plus rRNA), in green; coding sequences on the reverse strand, colored based on COG category; coding sequences on the forward strand, colored based on COG category; distribution of modified bases in the reverse strand (blue); distribution of modified bases in the forward strand (red); and genes associated with the restriction-modification system (in red).
![Figure 4. Circular genome map of the single chromosome of strain JXN-3, and color-coded list of Clusters of Orthologous Groups (COG) categories. Rings from inner to outer: %GC content, with GC content higher than average in green, and GC content lower than average in dark blue; GC skew [(G − C)/(G + C)], with positive in red and negative in purple; ncRNAs (tRNA plus rRNA), in green; coding sequences on the reverse strand, colored based on COG category; coding sequences on the forward strand, colored based on COG category; distribution of modified bases in the reverse strand (blue); distribution of modified bases in the forward strand (red); and genes associated with the restriction-modification system (in red).](/cms/asset/0197a9e5-66ee-427e-94a1-24972e3f41f6/tbeq_a_1808068_f0004_c.jpg)
Table 2. Genome statistics for strain JXN-3.
The ANI of a complete genomic sequence has been widely accepted as a boundary for species delineation [Citation48,Citation49]; an ANI of 97% corresponds to a DNA-DNA hybridization of 70% and a 16S rRNA sequence similarity of 98.65% [Citation50–52]. Other studies that identified strains as conspecific based on similar numbers have been performed, such as in Roseovarius albus sp., Agrobacterium bohemicum sp. and Agrobacterium deltaense sp. [Citation53–55]. The ANI between strain JXN-3 and M. saprophilus 32480 was 97%, identical to the ANI between strain JXN-3 and M. saprophilus NRRLB-24119 (). The ANIs between strain JXN-3 and other species of Mycetocola (i.e. M. lacteus JCM11654, M. miduiensis CGMCC1.11,101 M. manganoxydans CCTCCAB209002, M. reblochoni REB411 and M. tolaasinivorans IF016277) were 93%, 87%, 84%, 83% and 83%, respectively. Core and pan genomic analyses indicated that JXN-3 had 2536 core genes in common with M. saprophilus, but 2487, 2306, 1585, 1582 and 1567 core genes in common with M. lacteus, M. tolaasinivorans, M. miduiensis, M. manganoxydans and M. reblochoni respectively ().
Figure 5. Similarities between the genome of strain JXN-3 and those of strains of Mycetocola. (a) Average nucleotide identity (ANI) heatmap; (b) Venn diagram showing core gene overlap among tested strains.
Genome annotation and comprehensive analyses of strain JXN-3
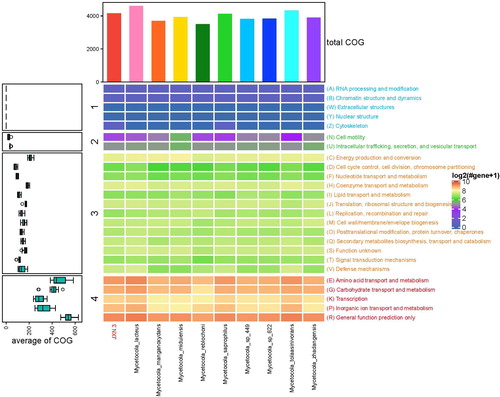
Of the 3094 identified protein CDS, 2967 were successfully annotated, with 2478 (80.09%) annotated in COG. The protein CDS fell into 24 COG categories. Seven genes potentially related to fungal cell wall degradation were identified based on COG annotations: two chitin-associated genes (chitin deacetylase, orf02111-1663, and chitinase, orf03713-2941) and four genes associated with glucan metabolism (1,4-beta-D-glucohydrolase, orf03144-2486; beta-glucanase, orf03320-2626; endoglucanase Acf2 orf00551-447, orf00553-448; and Exo-beta-1,3-glucanase, orf01977-1566). COG annotations of JXN-3 and M. lacteus, M. manganoxydans, M. miduiensis, M. reblochoni, M. saprophilus, Mycetocola sp. 449, Mycetocola sp. 622, M. tolaasinivorans and M. zhadangensis showed that JXN-3 and M. saprophilus are similar in genome function ().
Figure 6. Comprehensive analysis of genomic Clusters of Orthologous Groups (COG) annotations for JXN-3 and other members of the genus Mycetocola.
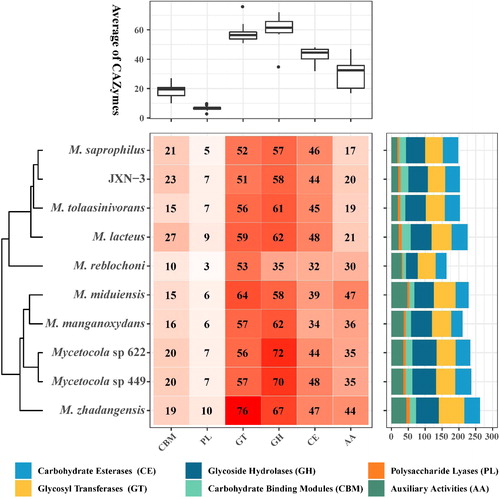
CAZymes, which play a key role in the breakdown of the cell wall, have been classified into six families: glycosyltransferases (GTs), glycoside hydrolases (GHs), polysaccharide lyases (PLs), carbohydrate esterases (CEs), carbohydrate-binding modules (CBMs) and auxiliary activities (AAs) [Citation56]. In this study, a total of 203 candidate CAZyme-encoding genes were identified in M. saprophilus JXN-3, including 58 (28.57%) GH, 51 (25.12%) GT, 44 (21.67%) CE, 23 (11.33%) CBM, 7 (3.45%) PL and 20 (9.85%) AA (). The total numbers and relative proportions of the CAZyme-encoding genes in JXN-3 were similar to those identified in other Mycetocola (). The CE, GH and PL families are collectively known as cell wall degrading enzymes (CWDEs), and are thought to help pathogens invade host cells [Citation57]. The presence of chitinase (GH23) [Citation58], chitin deacetylase (CE4) [Citation59] and chitin-binding enzymes (CBM2, CBM5, CBM12, CBM50) in the JXN-3 genome suggested that this strain may have chitin degradation abilities ().
Figure 7. The number of genes encoding carbohydrate-active enzymes (CAZymes) in JXN-3 and other members of the genus Mycetocola. AA: auxiliary activities; CE: carbohydrate esterases; PL: polysaccharide lyases; CBM: carbohydrate-binding modules; GT: glycosyl transferases; GH: glycoside hydrolases.
The stipe cell wall of F. velutipes is composed of chitin, glucan and small amounts of dispersed proteins and lipids; the cell wall acts as a barrier to pathogen invasion [Citation60,Citation61]. In F. velutipes, stipe extension activity occurs in the first 10–30 mm of the apical zone [Citation62]. Stipe extension may be mediated by endogenous expansin-like proteins, which promote cell wall expansion by destroying non-covalent bonds between glucan or chitin chains [Citation62]. In the early stage of JXN-3 infection, small brown spots were observed on the stipe 25–45 mm below the fungal cap, probably because this area was part of the fragile elongation zone. The sunken and water-soaked lesions subsequently observed on the stipes of F. velutipes during the pathogenic challenges indicated that the pathogenic strain degraded the fungal cell wall.
Secondary metabolite clusters in JXN-3
Our antiSMASH analysis indicated that the JXN-3 genome possessed five gene clusters known to synthesize secondary metabolites. These clusters included 69 genes: 27 associated with the production of the antifungal compounds bacteriocin, 23 associated with beta-lactone and 19 associated with terpene (Supplemental Table S3). A phylogenetic tree () was constructed using the single-copy orthologs from the bacteriocin clusters of JXN-3 and five other members of Mycetocola. These species formed four homologous gene clusters and one JXN-3-unique orthologous cluster (cluster 1). These genes may contribute to the pathogenesis of soft rot in F. velutipes stipes. Specifically, bacteriocins are small cationic peptides with broad-spectrum antimicrobial activity, which can inhibit cell wall biosynthesis [Citation63,Citation64]. Future studies should further investigate the involvement of these proteins in the incidence and progression of soft rot disease in F. velutipes. The tree depicts the relationships among JXN-3 and other members of Mycetocola.
Figure 8. Comparisons of bacteriocin clusters in JXN-3 and other members of the genus Mycetocola. Numbers at nodes represent bootstrap values based on 1000 replicates. The arrow represents the secondary metabolite (SM) gene arrangement and strand pattern.

Conclusions
M. saprophilus JXN-3 is able to grow at low temperatures (15 °C), similar to F. velutipes, and can cause soft rot in the stipes of F. velutipes. The sunken and water-soaked lesions on F. velutipes indicated that M. saprophilus JXN-3 degraded the fungal cell wall. In this study, we identified several genes encoding proteins associated with the synthesis of cell wall-degrading enzymes and antifungal compounds. Unlike other members of Mycetocola, JXN-3 possessed a unique gene cluster (cluster 1) that synthesized bacteriocin; this gene may inhibit cell wall synthesis. To our knowledge, this is the first implication of a M. saprophilus in the pathogenesis of soft rot in F. velutipes. The availability of the strain JXN-3 genome will support further investigations of the pathogenic mechanisms underlying soft rot disease, which may lead to innovations in soft rot disease control.
Supplemental Material
Download PDF (246.9 KB)Acknowledgements
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript. We gratefully acknowledge the Whole-Genome Sequencing of M. saprophilus JXN-3 performed by Sinotech Genomics Co., Ltd.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Tang C, Hoo PCX, Tan LTH, et al. Golden needle mushroom: a culinary medicine with evidenced-based biological activities and health promoting properties. Front Pharmacol. 2016;7:474. doi:10.3389/fphar.2016.00474
- Li X, Li Y. Quality comparison and analysis on white Flammulina velutipes grown with bottle lines in China. Edib Fungi China. 2014;32:20–24.
- Wu Z, Peng W, He X, et al. Mushroom tumor: a new disease on Flammulina velutipes caused by Ochrobactrum pseudogrignonense. FEMS Microbiol Lett. 2016;363(2):fnv226. doi:10.1093/femsle/fnv226
- Liu K, Zhang G, Ma H, et al. Major infectious diseases and its preventive measures of Flammulina velutipe. Edib Fungi China. 2018;37:78–81. doi:10.13629/j.cnki.53-1054.2018.04.018
- Bessette AE, Kerrigan RW, Jordan DC. Yellow blotch of Pleurotus ostreatus. Appl Environ Microbiol. 1985;50(6):1535–1537. doi:10.1128/AEM.50.6.1535-1537.1985
- Thorn G, Tsuneda A. Molecular genetic characterization of bacterial isolates causing brown blotch on cultivated mushrooms in Japan. Mycoscience. 1996;37(4):409–416. doi:10.1007/BF02460997
- Chowdhury PR, Pay J, Braithwaite M. Isolation, identification and ecology of Ewingella americana (the causal agent of internal stipe necrosis) from cultivated mushrooms in New Zealand. Aust Plant Pathol. 2007;36(5):424–428. doi:10.1071/AP07045
- Lee CJ, Jhune CS, Cheong JC, et al. Occurrence of internal stipe necrosis of cultivated mushrooms (Agaricus bisporus) caused by Ewingella americana in Korea. Mycobiology. 2009;37(1):62–66. doi:10.4489/MYCO.2009.37.1.062
- Han HS, Jhune CS, Cheong JC, et al. Occurrence of black rot of cultivated mushrooms (Flammulina velutipes) caused by Pseudomonas tolaasii in Korea. Eur J Plant Pathol. 2012;133(3):527–535. doi:10.1007/s10658-012-9941-4
- Lincoln SP, Fermor TR, Tindall BJ. Janthinobacterium agaricidamnosum sp. nov, a soft rot pathogen of Agaricus bisporus. Int J Syst Evol Microbiol. 1999;49(4):1577–1589. doi:10.1099/00207713-49-4-1577
- Gill WM, Tsuneda A. The interaction of the soft rot bacterium Pseudomonas gladioli pv. agaricicola with Japanese cultivated mushrooms. Can J Microbiol. 1997;43(7):639–648. doi:10.1139/m97-091
- Lee CJ, Yun HS, Jhune CS, et al. Occurrence of bacterial soft rot of Pleurotus ostreatus caused by Burkholderia gladioli pv. agaricicola in Korea. J Plant Pathol. 2010;92:235–240.
- Okamoto H, Sato M, Isaka M. Bacterial soft rot of winter mushroom and oyster mushroom caused by Erwinia carotovora subsp. carotovora. Jpn J Phytopathol. 1999;65(4):460–464. doi:10.3186/jjphytopath.65.460
- Kim MK, Lee SH, Lee YH, et al. Characterization and chemical control of soft rot disease caused by Pantoea sp. strain PPE7 in Pleurotus eryngii mushroom crops. Eur J Plant Pathol. 2015;141(2):419–425. doi:10.1007/s10658-014-0538-y
- Liu JY, Men JL, Chang MC, et al. iTRAQ-based quantitative proteome revealed metabolic changes of Flammulina velutipes mycelia in response to cold stress. J Proteom. 2017;156:75–84. doi:10.1016/j.jprot.2017.01.009
- Cho H, Park JY, Kim YK, et al. Whole-genome sequence of Erwinia persicina b64, which causes pink soft rot in onions. Microbiol Resour Announc. 2019;8(1):e01302. doi:10.1128/MRA.01302-18
- Ma X, Perna NT, Glasner JD, et al. Complete genome sequence of Dickeya dianthicola ME23, a pathogen causing blackleg and soft rot diseases of potato. Microbiol Resour Announc. 2019;8(7):18. doi:10.1128/MRA.01526-18
- Fang Z. Research methods of plant disease. Beijing: China Agric Press; 1998.
- Bavykin SG, Lysov YP, Zakhariev V, et al. Use of 16S rRNA, 23S rRNA, and gyrB gene sequence analysis to determine phylogenetic relationships of Bacillus cereus group microorganisms. J Clin Microbiol. 2004;42(8):3711–3730. doi:10.1128/JCM.42.8.3711-3730.2004
- Richert K, Brambilla E, Stackebrandt E. Development of PCR primers specific for the amplification and direct sequencing of gyrB genes from microbacteria, order Actinomycetales. J Microbiol Methods. 2005;60(1):115–123. doi:10.1016/j.mimet.2004.09.004
- Richert K, Brambilla E, Stackebrandt E. The phylogenetic significance of peptidoglycan types: molecular analysis of the genera Microbacterium and Aureobacterium based upon sequence comparison of gyrB, rpoB, recA and ppk and 16SrRNA genes. Syst Appl Microbiol. 2007;30(2):102–108. doi:10.1016/j.syapm.2006.04.001
- Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi:10.1016/S0022-2836(05)80360-2
- Rozewicki J, Li S, Amada KM, et al. MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res. 2019;47(W1):W5–W10. doi:10.1093/nar/gkz342
- Liu K, Linder CR, Warnow T. RAxML and FastTree: comparing two methods for large-scale maximum likelihood phylogeny estimation. PLoS One. 2011;6(11):e27731. doi:10.1371/journal.pone.0027731
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39(4):783–791. doi:10.1111/j.1558-5646.1985.tb00420.x
- Chin CS, Alexander DH, Marks PK, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10(6):563–569. doi:10.1038/nmeth.2474
- Sommer DD, Delcher AL, Salzberg SL, et al. Minimus: a fast, lightweight genome assembler. BMC Bioinf. 2007;8(1):64. doi:10.1186/1471-2105-8-64
- Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20(16):2878–2879. doi:10.1093/bioinformatics/bth315
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–964. doi:10.1093/nar/25.5.955
- Lagesen K, Hallin P, Rødland EA, et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–3108. doi:10.1093/nar/gkm160
- Nawrocki EP, Burge SW, Bateman A, et al. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 2015;43(Database issue):D130–D137. doi:10.1093/nar/gku1063
- Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 2009;25:4.10. doi:10.1002/0471250953.bi0410s25
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–580. doi:10.1093/nar/27.2.573
- Bland C, Ramsey TL, Sabree F, et al. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics. 2007;8:209. doi:10.1186/1471-2105-8-209
- Thiel T, Michalek W, Varshney R, et al. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet. 2003;106(3):411–422. doi:10.1007/s00122-002-1031-0
- Flusberg BA, Webster DR, Lee JH, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods. 2010;7(6):461–465. doi:10.1038/nmeth.1459
- Zhu L, Liu Q, Liu H, et al. Mycetocola miduiensis sp. nov, a psychrotolerant bacterium isolated from Midui glacier. Int J Syst Evol Microbiol. 2013;63(Pt 7):2661–2665. doi:10.1099/ijs.0.047985-0
- Bora N, Vancanneyt M, Gelsomino R, et al. Mycetocola reblochoni sp. nov, isolated from the surface microbial flora of Reblochon cheese. Int J Syst Evol Microbiol. 2008;58(Pt 12):2687–2693. doi:10.1099/ijs.0.64201-0
- Doroghazi JR, Albright JC, Goering AW, et al. A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat Chem Biol. 2014;10(11):963–968. doi:10.1038/nchembio.1659
- Tsukamoto T, Takeuchi M, Shida OM, et al. Proposal of Mycetocola gen. nov. in the family Microbacteriaceae and three new species, Mycetocola saprophilus sp. nov, Mycetocola tolaasinivorans sp. nov. and Mycetocola lacteus sp. nov, isolated from cultivated mushroom, Pleurotus ostreatus. Int J Syst Evol Microbiol. 2001;51(Pt 3):937–944. doi:10.1099/00207713-51-3-937
- Shen L, Liu Y, Yao T, et al. Mycetocola zhadangensis sp. nov., isolated from snow. Int J Syst Evol Microbiol. 2013;63(Pt 9):3375–3378. doi:10.1099/ijs.0.047159-0
- Luo X, Wang J, Zeng XC, et al. Mycetocola manganoxydans sp. nov, an actinobacterium isolated from the Taklamakan desert. Int J Syst Evol Microbiol. 2012;62(Pt 12):2967–2970. doi:10.1099/ijs.0.038877-0
- Yi H, Cho YJ, Yoon SH, et al. Comparative genomics of Neisseria weaveri clarifies the taxonomy of this species and identifies genetic determinants that may be associated with virulence. FEMS Microbiol Lett. 2012;328(2):100–105. doi:10.1111/j.1574-6968.2011.02485.x
- Tatusov RL, Fedorova ND, Jackson JD, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi:10.1186/1471-2105-4-41
- Zhang H, Yohe T, Huang L, et al. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018;46(W1):W95–W101. doi:10.1093/nar/gky418
- Blin K, Shaw S, Steinke K, et al. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019;47(W1):W81–W87. doi:10.1093/nar/gkz310
- Nguyen LT, Schmidt HA, von Haeseler A, et al. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32(1):268–274. doi:10.1093/molbev/msu300
- Goris J, Konstantinidis KT, Klappenbach JA, et al. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57(Pt 1):81–91. doi:10.1099/ijs.0.64483-0
- Konstantinidis KT, Ramette A, Tiedje JM. The bacterial species definition in the genomic era. Philos Trans R Soc Lond B Biol Sci. 2006;361(1475):1929–1940. doi:10.1098/rstb.2006.1920
- Grim CJ, Kotewicz ML, Power KA, et al. Pan-genome analysis of the emerging foodborne pathogen Cronobacter spp. suggests a species-level bidirectional divergence driven by niche adaptation. BMC Genomics. 2013;14:366. doi:10.1186/1471-2164-14-366
- Haley BJ, Grim CJ, Hasan NA, et al. Comparative genomic analysis reveals evidence of two novel Vibrio species closely related to V. cholerae. BMC Microbiol. 2010;10(1):154. doi:10.1186/1471-2180-10-154
- Kim M, Oh HS, Park SC, et al. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol. 2014;64(Pt_2):346–351. doi:10.1099/ijs.0.059774-0
- Lucena T, Ruvira MA, Macián MC, et al. Roseovarius albus sp. nov, a new Alphaproteobacterium isolated from the Mediterranean Sea. Antonie Van Leeuwenhoek. 2014;105(4):671–678. doi:10.1007/s10482-014-0121-8
- Zahradník J, Nunvar J, Pařízková H, et al. Agrobacterium bohemicum sp. nov. isolated from poppy seed wastes in central Bohemia. Syst Appl Microbiol. 2018;41(3):184–190. doi:10.1016/j.syapm.2018.01.003
- Yan J, Li Y, Han XZ, et al. Agrobacterium deltaense sp. nov, an endophytic bacteria isolated from nodule of Sesbania cannabina. Arch Microbiol. 2017;199(7):1003–1009. doi:10.1007/s00203-017-1367-0
- Lombard V, Golaconda Ramulu H, Drula E, et al. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(Database issue):D490–D495. doi:10.1093/nar/gkt1178
- Ospina-Giraldo MD, Griffith JG, Laird EW, et al. The CAZyome of Phytophthora spp.: a comprehensive analysis of the gene complement coding for carbohydrate-active enzymes in species of the genus Phytophthora. BMC Genomics. 2010;11:525. doi:10.1186/1471-2164-11-525
- Takao A, Noriko K, Shoko S, et al. Crystal structures of the catalytic domain of a novel glycohydrolase family 23 chitinase from Ralstonia sp. A-471 reveals a unique arrangement of the catalytic residues for inverting chitin hydrolysis. J Biol Chem. 2013;288(26):18696–18706. doi:10.1074/jbc.M113.462135
- Park BH, Karpinets TV, Syed MH, et al. CAZymes analysis toolkit (CAT): web service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiology. 2010;20(12):1574–1584. doi:10.1093/glycob/cwq106
- Mol PC, Vermeulen CA, Wessels JGH. Diffuse extension of hyphae in stipes of Agaricus bisporus may be based on a unique wall structure. Mycol Res. 1990;94(4):480–488. doi:10.1016/S0953-7562(10)80007-3
- Kamada T, Takemaru T, Prosser JI, et al. Right and left handed helicity of chitin microfibrils in stipe cells in Coprinus cinereus. Protoplasma. 1991;165(1-3):64–70. doi:10.1007/BF01322277
- Fang H, Zhang W, Niu X, et al. Stipe wall extension of Flammulina velutipes could be induced by an expansin-like protein from Helix aspersa. Fungal Biol. 2014;118(1):1–11. doi:10.1016/j.funbio.2013.10.003
- Fons M, Gomez A, Karjalainen T. Mechanisms of colonisation and colonisation resistance of the digestive tract part 2: bacteria/bacteria interactions. Microb Ecol Health Dis. 2000;12:240–246. doi:10.1080/089106000750060495
- Perez RH, Zendo T, Sonomoto K. Novel bacteriocins from lactic acid bacteria (LAB): various structures and applications. Microb Cell Fact. 2014;13(Suppl 1):S3. doi:10.1186/1475-2859-13-S1-S3