
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Correctly identifying the protein-ATP binding site is valuable for both protein function annotation and new drug discovery. However, the number of non-ATP-binding residues is much more than the number of ATP-binding residues, which makes the prediction a classical imbalanced learning problem. Previous studies often apply the under-sampling technique to construct a relatively balanced dataset, but some information is inevitably lost during the sample process. In this work, we utilize the SMOTE algorithm, which generates the balanced dataset by generating ATP-binding sites with the idea of interpolation. The Random Forest is selected as classifier to ensure the acceptable training speed. With the combination of complementary template-based method, the prediction performance of the proposed method is further improved. After comparing with other sequence-based predictors, our proposed method achieves satisfying performance and proved to be efficient for ATP-binding sites prediction.
Introduction
Adenosine-5′-triphosphate (ATP) is an important nucleotide which is involved in various biological processes in cells such as energy metabolism [Citation1], membrane transportation [Citation2], signaling [Citation3] and replication of DNA [Citation4]. As of February 2019, 6885 structures in the Protein Data Bank (PDB) [Citation5] are annotated as ATP-binding, which accounts for about 4.63% among all records. The ATP-binding proteins contain ATP-binding sites which allow the interaction between ATP and proteins. Therefore, accurately identifying the ATP-binding sites in proteins is of great importance for protein function detection [Citation6, Citation7] and new drug development of antibacterial and anti-cancer chemotherapy[Citation8–10].
Previous efforts to identify the ATP-binding sites in proteins have been made using wet-lab techniques. However, the traditional experimental methods are often cost-intensive and time-consuming, which makes them insufficient for wide application. Based on this background, computational methods which combine the machine learning technique and biological information has been developed to predict the location of various binding sites [Citation11–15]. Two types of biological information are often involved in the prediction of ATP-binding sites: sequence information and structural information. However, until February 2019, the number of known protein sequences collected in Swiss-Prot database [Citation16] which are manually annotated was about 559077, whereas the number of known structures stored in PDB database was merely about 148827. The huge gap between the number of accessions corresponding to the two types of biological information has revealed the wider application prospect of predicting the ATP-binding sites based on sequence information. Therefore, many pioneering works have been proposed in recent years. In 2009, Chauhan et al. [Citation17] introduced ATPint, which is the first custom-designed predictor for ATP-binding sites in protein sequences based on a dataset consisting of 168 non-redundant ATP-binding proteins. The position specific scoring matrix (PSSM) is used to represent the evolutionary feature and the support vector machine (SVMs) is used to obtain the prediction result. In 2011 and 2012, Chen et al. [Citation18, Citation19] developed two stronger predictors called ATPsite and NsitePred, respectively, based on a larger-scale dataset. Besides the PSSM, a number of new features are applied in these methods such as the residue conservation scores, the predicted secondary structure and collocation of amino acid pairs, which makes them obtain better prediction accuracies. Yu et al. [Citation20] developed the TargetATPsite in 2013 with the idea of classifier ensemble, and successfully improved the prediction performance. In 2015, Andrews et al. [Citation21] developed TSC_ATP, which is a two-layer structure algorithm for the prediction of ATP-binding sites from protein sequences.
Although significant progress has been made by these previous studies, there is still place for performance improvement. The prediction of ATP-binding sites in protein sequences is a typical imbalanced classification problem, since the number of non-ATP-binding residues is much more than the number of ATP-binding residues in protein sequences. For example, in the dataset of ATP227, which was applied in Chen’s [Citation18] work, the number of ATP-binding residues is 3393, whereas the number of non-ATP-binding residues is 80409. Therefore, directly using the traditional machine learning technique may lead to a biased result and poor performance. Previous works often utilized the under-sampling technique, which randomly selects the same number of samples from a majority class as the number in the minority class to construct a relatively balanced data subset that can solve the imbalanced problem [Citation17–21]. However, some valuable properties can be hidden in the non-ATP-binding residues; simply abandoning them will leave the training process incomplete and will have a negative effect on the prediction performance. In 2002, Chawla et al. [Citation22] introduced the algorithm of synthetic minority over-sampling technique (SMOTE) to solve the imbalanced learning problem from another aspect. The SMOTE algorithm does not abandon any sample in the dataset as it creates new samples with the idea of interpolation for the minority class. Compared with the under-sampling technique, the SMOTE algorithm keeps the complete training set without losing any valuable information, which helps the classifier receive more training information and keep more stable performance during the testing process [Citation23]. Compared with simply over-sampling, which just duplicates the minority samples, the SMOTE algorithm generates new minority samples based on interpolation, which efficiently increases the generalization capability and avoids the over-fitting problem for the classifier [Citation24].
In this study, we apply the SMOTE algorithm in combination with the Random Forest classification algorithm to predict the ATP-binding sites based on the information extracted from protein sequences. First, a balanced dataset is constructed by SMOTE; then, various sequence features are extracted and the random forest classifier is built based on these features; finally, in order to further improve the prediction performance, a complementary template-based method is applied, which is integrated with the Random Forest classifier to obtain the final decision. The five-fold cross-validation on two benchmark datasets has shown that our proposed method efficiently improves the prediction performance and provides insights into the mechanism of protein–ATP interaction.
Materials and methods
Datasets
Two benchmark datasets are applied to verify the effectiveness of our proposed method. The two datasets are constructed by previous studies and have been commonly used in other prediction methods.
ATP168: The dataset was originally constructed in the study of ATPint [Citation17]. Briefly, 360 ATP binding proteins are extracted from SuperSite encyclopedia [Citation25]. Then the CD-HIT [Citation26] is applied to remove the redundant protein sequences and reduce the sequence identity to 40%. Finally, the dataset is formed with 168 non-redundant ATP-binding proteins which contains 3,104 ATP-binding residues and 59,225 non-ATP-binding residues.
ATP227: The dataset was firstly curated in the study of ATPsite [Citation18] by Kurgan. The maximum pairwise sequence identity of proteins is reduced to 40% by CD-HIT. A residue is defined as ATP-binding residue if any of its non-hydrogen atoms is less than 3.9 Å away from any non-hydrogen atom in the ATP molecule. Finally, the dataset is constructed with 227 non-redundant ATP binding proteins and contains 3,393 ATP-binding residues and 80,409 non-ATP-binding residues.
Primary sequential feature-based predictor
The problem of imbalanced learning
The prediction of ATP-binding sites is a classical imbalanced learning problem, since the number of non-binding residues is far more than the number of binding residues. The imbalance between the two classes often leads to biased prediction performance as the classifier is not sensitive enough for binding residues. In order to avoid this situation, previous studies tend to apply the under-sampling technique which randomly extracts a similar number of non-binding residues and combines them with binding residues to construct a relatively balanced dataset. However, the under-sampling technique will inevitably lose some non-binding residues which are equally important for the classifier because some valuable binding properties are hidden in these non-binding residues. In this study, the SMOTE algorithm is utilized to solve the imbalanced learning problem which will not lose any information in the dataset. For each binding residue, the SMOTE algorithm first randomly selects a neighboring binding residue according to the distance from the feature matrix, for example the Euclidean distance. Then a straight line is drawn to connect the binding residue and its neighboring binding residue. Finally, a new binding residue sample will be generated by selecting a point in the straight line based on the idea of interpolation. Compared with traditional over-sampling which increases the number of minority class by simply duplicating samples, SMOTE algorithm has the advantage that the classifier can be more general and efficiently avoid being over-fitted. The formula of generating new binding residue sample for SMOTE algorithm is shown as follows:
Where stands for the new-generated binding residue sample,
stands for the binding residue in the training set and
stands for the neighboring binding residues of
Feature representation
In this study, we utilize a sliding window of size 15 including the centered residue and 7 adjacent residues on the left and right side to represent the feature pattern. The size of the sliding window is determined by experimental attempts. We tried different sizes for the sliding window from 1 to 19, the Matthews Correlation Coefficient (MCC) values corresponding to different sizes of sliding window on two benchmark datasets are shown in .
Figure 1. MCC values for different sizes of sliding window on ATP-168 and ATP-227.
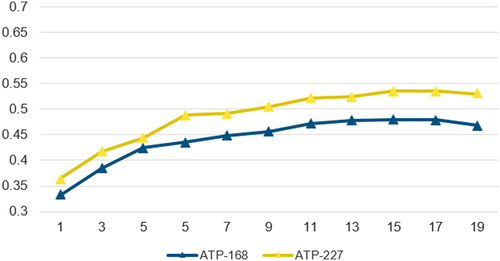
After determining the size of the sliding window, these features are considered to construct the feature matrix including:
Position Specific Scoring Matrix (PSSM)
Position Specific Scoring Matrix (PSSM) well encodes evolutionary information of a protein sequence. Previous studies have shown its significance in ligand binding sites prediction and structure prediction, such as protein-RNA binding site prediction and protein secondary structure prediction [Citation20, Citation27].
For a query sequence, the PSSM is generated by running PSI-BLAST [Citation28] to search against the Swiss-Port database with three iterations and E-value of 0.001. Then, we normalize the original values in the PSSM to the range of 0 and 1 with logistic function as follows:
Where the
represents the original value in the PSSM and the
Predicted secondary structure
Previous studies have shown that secondary structures are relevant to ATP-binding residues; different secondary structure composition is noticed in ATP-binding residues and non-ATP-binding residues. In this study, the PSIPRED [Citation27] software is applied to provide the secondary structure information which predicts the probabilities belonging to three types of secondary structure including coil (C), helix (H) and strand (E) for each residue based on protein sequences. Therefore, for a sliding window of size 15, the dimension of predicted secondary structure feature is 3*15 = 45.
Predicted solvent accessibility
The value of solvent accessibility represents the surface area of a given residue that is accessible to the solvent. A residue with higher solvent accessibility often means that it has more potential to interact with molecules in an outer environment. In this study, the ASAquick [Citation29] is utilized to predict the solvent accessibility with sequence information. Therefore, the dimension of solvent accessibility feature for the sliding window is 1*15 = 15.
Physicochemical property
According to the dipoles and volumes of side chains of amino acids, 20 types of amino acids can be divided into 7 classes as follows: class a = {Ala, Gly, Val}, class b = {Ile, Leu, The, Pro}, class c = {His, Asn, Gln, Trp}, class d = {Tyr, Met, Thr, Ser}, class e = {Arg, Lys}, class f = {Asp,Glu}, class g = {Cys}. Therefore, a 7-dimensional one-hot binary key is used to encode the physicochemical property for each residue. For the sliding window of size 15, the dimension of physicochemical property is 7*15 = 105.
In summary, for each residue in a protein sequence, four types of biological information are extracted including PSSM (300), predicted secondary structure (45), predicted solvent accessibility (15) and physicochemical property (105). The dimension of the feature matrix is 300 + 45 + 15 + 105 = 465.
Random Forest classifier
The Random Forest (RF) algorithm has been successfully applied in many fields for data classification and achieves satisfying performance. RF uses an ensemble of unpruned decision trees, each of which is built from a bootstrap sample of the training data using a randomly selected subset of variables. It is worth mentioning that after utilizing the SMOTE algorithm, the scale of the dataset has increased, which means that the training process will become more time consuming. Based on this background, the RF classification algorithm is selected as our classifier. The reason is that for large-scale of high-throughput training data, the RF classifier shows better training speed than other classifiers, for example the support vector machines (SVM). After the SMOTE process, the training speed of the RF classifier for ATP-168 dataset is about five minutes, whereas the training speed of SVM is more than ten hours. Therefore, in order to solve the imbalanced learning problem and make our method more practicable, the combination of SMOTE algorithm and RF classifier is used to build the primary feature-based ATP-binding sites predictor.
Complementary template-based method
Inspired by previous methods for RNA-binding site prediction [Citation30, Citation31], we also develop a template-based prediction method as complement to improve the prediction performance. The template-based method detects the sequence homology between the query sequence and sequences in the training set. The small homologous segments in the query sequence probably have similar functions to the segments in the training data, which are called the templates. In this study, the templates are detected using the bit-score provided by the PSI-BLAST program. It is worth mentioning that even though the sequence pairwise identity is less than 40% among the dataset, small similar segments still exist which can be used for template detection. If the bit-score between the query sequence and one sequence in the training set is larger than a threshold, we will check the template in the training set to figure out whether it contains an ATP-binding site or not. If the binding site is detected, the corresponding residue in the similar segment from the query sequence will be labeled as ATP-binding. Otherwise, if the ATP-binding site is not included in the template or there is no template in the training set, the complementary template-based method will not be applied. The bit-score threshold in the proposed method is set to 50.
Architecture of the proposed method
The architecture of the proposed method is shown in . The training process of the primary sequential feature-based predictor is shown in green lines; the testing process of the primary sequential feature-based predictor is shown in blue lines and the process of complementary template-based predictor is shown in orange lines. For a query protein sequence, PSI-BLAST is used to generate the PSSM profile, PSIPRED is applied to generate the predicted secondary structure and ASAquick predicts the solvent accessibility. The physicochemical feature is directly calculated according to the composition of protein sequence and is combined with these inputs to construct the feature matrix. Then the feature matrix is sent into the RF classifier which is trained with a relatively balanced dataset constructed by the SMOTE algorithm to generate the probability of ATP-binding. Finally, the complementary template-based method is applied to detect the ATP-binding site from segment homology aspect. The final prediction result is obtained by combining the output of the RF classifier and template-based method. The corresponding framework of the prediction procedure is shown in .
Figure 2. Architecture of the proposed method.
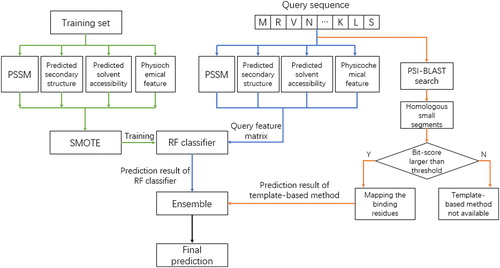
Figure 3. Framework of the proposed prediction procedure.

Results and discussions
Performance evaluation indices
In this study, five-fold cross-validation is used to assess the prediction performance of the proposed method. First, all the residues in the dataset are randomly divided into five parts. Then four parts are used to train the classifier and the remaining part is used to examine the prediction performance. Finally, this procedure is repeated five times to make sure that each part has been applied for testing. The final prediction performance will be obtained by averaging the prediction performance of five parts.
Four routinely used evaluation indices are selected to show the effectiveness of our proposed method including the overall accuracy (ACC), sensitivity (Sen), specificity (Spe) and the Matthews Correlation Coefficient (MCC), which is one of the most potent evaluation criteria for the binary classifier. These evaluation indices can be defined as follows:
where the TP, TN, FP and FN represent the number of true positive, true negative, false positive and false negative samples, respectively. However, these four indices are threshold-dependent, which means they are influenced by the threshold of prediction. The reason is that for a soft-type predictor which outputs a continuous numeric value to represent the probability of a sample belonging to a predicted class, applying different prediction thresholds will produce different prediction confusion matrices, which leads to different values for the evaluation indices. How to objectively report these indices is an important problem. For the situation of imbalanced learning, overly pursuing the high overall accuracy is not appropriate and may lead to deceiving prediction performance as the number of the majority class greatly exceeds the number of the minority class. Thus, in order to objectively show the prediction performance, the evaluation index of AUC is introduced, which is the area under the Receiver Operating Characteristic (ROC) curves. The introduction of AUC, which is threshold-independent, increases in direct proportion to the prediction performance.
For threshold-dependent evaluation indices, inspired by the work of ATPsite and NsitePred, the threshold that maximizes the prediction’s MCC value is selected to report the prediction performance.
SMOTE algorithm helps to improve the prediction performance
As severe imbalance exists between the ATP-binding residues and non-binding residues, in this study, we introduce the SMOTE algorithm to the problem of ATP-binding prediction instead of under-sampling that has been commonly used in previous studies. Compared with under-sampling, the SMOTE algorithm will not lose any training information and constructs the balanced dataset using manually generated samples instead of simply duplicating the minority samples, which efficiently avoids over-fitting for the classifier. In order to show the effectives of the SMOTE algorithm in ATP-binding prediction, for two benchmark datasets, we utilize under-sampling, original dataset without under-sampling and the SMOTE algorithm, respectively, to construct the RF predictors. The prediction performance of the RF classifier over five-fold cross-validation is shown in .
Table 1. Prediction performance based on the under-sampled dataset, original dataset and SMOTE dataset over five-fold cross-validation.
According to , the classifier constructed on datasets applied with the SMOTE algorithm achieves the best performance among three predictors, with the highest ACC of 0.959 and MCC of 0.480 on ATP-168, highest ACC of 0.969 and MCC of 0.535 on ATP-227 respectively. By comparing the performance of the under-sampling technique and SMOTE algorithm, we can find that the sensitivity and specificity are improved on both datasets, which means the classifier has better distinguishing capacity on both ATP-binding residues and non-binding residues. Since the datasets after SMOTE and under-sampling are relatively balanced, the improvements can be explained by: first, the dataset of under-sampling does not contain all the non-binding residues which are equally valuable for prediction performance; second, the SMOTE algorithm generates the new minority samples based on random linear interpolation, which makes the newly generated samples as significant as the samples in the original dataset for prediction. When comparing the performance of the SMOTE algorithm and the original data, the specificity of the dataset after the SMOTE algorithm is slightly lower than that of the original dataset. This can be explained by the severe imbalance of the original dataset: it contains much more non-binding residues than ATP-binding residues. Therefore, the classifier is biased toward non-binding residues. However, for ATP-binding residues, the sensitivity of the dataset after the SMOTE algorithm greatly surpasses the sensitivity of the original dataset, which is 0.128 and 0.131 higher on ATP-168 and ATP-227, respectively. The improvements have shown that the classifier based on datasets applied with the SMOTE algorithm has better ability to identify the ATP-binding residues in protein sequences.
However, these four indices are threshold-dependent, in order to further demonstrate the effectiveness of the SMOTE algorithm on ATP-binding site prediction, we draw the ROC curves to compare the prediction performance of classifiers constructed on datasets applied with SMOTE algorithm, original datasets without under-sampling and datasets with under-sampling. shows the prediction ROC curves on ATP-168 and ATP-227.
Figure 4. Prediction ROC curves of datasets with SMOTE algorithm, original datasets without under-sampling and datasets with under-sampling on ATP-168 (a) and ATP-227 (b).
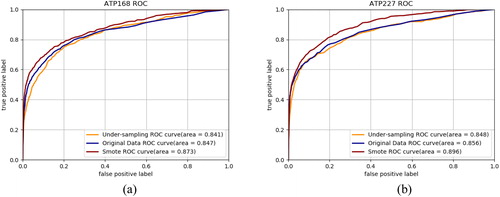
From , it can be found that the classifier on SMOTE datasets outperforms the classifiers on the under-sampled dataset and the original dataset on both ATP-168 and ATP-227. The AUC value, which is threshold-independent, is calculated on both datasets. For ATP-168, the AUC for SMOTE, original dataset and under-sampling are 0.873, 0.847 and 0.841, respectively. For ATP-227, the AUC for SMOTE, original dataset and under-sampling are 0.896, 0.856 and 0.848, respectively. The best AUC values for both datasets are obtained by the classifier with the SMOTE algorithm, which is coincident with the result of threshold-dependent indices.
Based on the performance comparison on both threshold-dependent and threshold-independent evaluation indices, the SMOTE algorithm that we applied to train our prediction classifier can efficiently restrain the negative effect from data-imbalance on ATP-binding site prediction. Besides, it can further improve the prediction performance as compared with the under-sampling technique.
Effectiveness of the complementary template-based method
Inspired by Yang’s work in predicting the RNA-binding sites, we develop a complementary template-based method for ATP-binding site prediction. The method is described in the Materials and methods section. For five-fold cross-validation, four parts are used for training and the remaining part is used for testing, the template will be searched from four training parts. In order to evaluate the usefulness of the complementary template-based method, we compare the prediction performance with and without the template-based method on both ATP-168 and ATP-227 datasets. The prediction performance over five-fold cross-validation is shown in .
Table 2. Comparison of prediction performance with and without the template-based method on ATP-168 and ATP-227 over five-fold cross-validation.
It can be observed that the MCC value improves by 0.029 and 0.024 on ATP-168 and ATP-227, respectively, after appending the complementary template-based method. Besides the MCC value, the sensitivity also improves by 0.042 and 0.044 for both datasets, which demonstrates that the complementary template-based method indeed helps to identify the ATP-binding residues in the protein sequence. Similar to the validation process for the SMOTE algorithm, we also examine the effectiveness of our complementary template-based method with threshold-independent indices. shows the ROC curves of the prediction result with and without the template-based method on both ATP-168 and ATP-227 datasets.
Figure 5. ROC curves of prediction result with and without template-based method on ATP-168 (a) and ATP-227 (b).
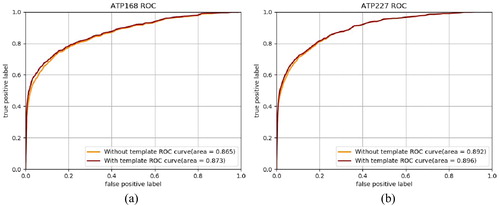
We can find that the prediction performance with the complementary template-based method is better than the performance without the template-based method. On ATP-168, the AUC of the prediction with the template-based method is 0.873, whereas the AUC without the template-based method is 0.865. On ATP-227, the AUC values with and without the template-based method are 0.896 and 0.892. The improvements on both threshold-dependent and threshold-independent evaluation indices have shown that the combination of the primary feature-based method and the complementary template-based method achieves better performance compared to the single prediction method. This indicates that the combination strategy is efficient for ATP binding-site prediction.
Performance comparison with previous works
Comparison with ATPint
ATPint is the first prediction method which was specifically designed for protein-ATP binding sites proposed by Chauhan et al. [Citation17]. ATPint utilizes evolutionary features extracted from protein sequential information and SVM as classifier for prediction. Chauhan et al. [Citation17] tried a number of thresholds to reveal the prediction performance under different thresholds and finally the threshold whose sensitivity and specificity was nearly equal was selected to report the performance. In order to fairly compare the prediction performance with ATPint, we also adjusted our threshold to balance the sensitivity and specificity similar to the process of ATPint. The ATP-168 dataset was created in the work of ATPint; therefore, we performed five-fold cross-validation on ATP-168 to compare with ATPint. The comparison result is shown in .
Table 3. Performance comparison of our proposed method with ATPint on ATP168 over five-fold cross-validation.
From , it is easy to observe that all of four threshold-dependent evaluation indices are improved. The ACC improves from 0.751 of ATPint to 0.790 of proposed method and the MCC improves from 0.249 to 0.291. For AUC, which is threshold-independent, our proposed method still obtains a better performance of 0.873 over ATPint (0.823). The higher value in MCC demonstrates that our proposed method has better performance than ATPint under a suitable threshold, whereas the higher value in AUC indicates that our method obtains overall better performance than ATPint on both ATP-binding residues and non-ATP-binding residues regardless of which threshold is selected.
Comparison with other sequence-based methods
Chen et al. [Citation18] created the dataset of ATP-227 in their work of ATPsite. Since then, ATP-227 dataset is widely used as benchmark dataset for ATP-binding site prediction. Chen et al. [Citation19] also developed another prediction method called NsitePred, which utilizes the sequential information combined with predicted structural information. Yu et al. [Citation32] developed the TargetATP, which combines Adaboost and SVM to predict the ATP-binding sites. Then they developed another prediction method named TargetATPsite [Citation20], which utilizes the image sparse representation technique to encode the evolutionary information for prediction. Andrews and Hu [Citation21] developed a two-stage prediction method called TSC_ATP with the structure of two-layered K-Nearest Neighbors (K-NN) classifier. These mentioned methods all utilize the under-sampling technique to solve the imbalanced data problem. In order to fairly compare with them, we also made prediction on ATP-227 over five-fold cross-validation. The prediction results comparison is shown in .
Table 4. Prediction results comparison of our proposed method with other sequence-based methods on ATP-227 over five-fold cross-validation.
As shown in , for threshold-dependent evaluation indices, our proposed method achieves the highest ACC and MCC of 0.969 and 0.535, which outperforms other sequence-based prediction methods in the table. At the same time, for threshold-independent evaluation indices, the AUC of our proposed method is 0.896, which is also better than those of the other methods. The result of the performance comparison demonstrates that our proposed method keeps outstanding and stable performance among other sequence-based predictors. The improvement can be explained from two aspects: first, the training set keeps the full information and solves the imbalanced problem by the SMOTE algorithm, whereas other sequence-based methods sacrifice some valuable information to keep the dataset balanced. Second, besides the classifier trained by the SMOTE algorithm, a template-based method is developed to further improve the prediction performance from homologous segments in the sequence.
In order to further prove the generalization ability of our proposed method, an independent testing set constructed by Chen et al. [Citation19] is applied. The independent testing set consists of 17 ATP-binding protein chains which share less than 40% sequence identity with protein chains in ATP-227. This process keeps the testing set strictly independent and can be correctly used to reveal the prediction performance. Based on this background, we train our predictor on the ATP-227 dataset and the independent testing set is used to evaluate the generalization ability of our proposed method. lists the performance comparison of our proposed method and other sequence-based methods on the independent testing set.
Table 5. Performance comparison of our proposed method with other sequence-based methods on an independent testing set.
It can be found from that our proposed method obtains the AUC of 0.912, which is equal to TargetATP but much better than the other predictors. However, the MCC and ACC of our proposed method are 0.553 and 0.973, respectively, which are higher than those of TargetATP (equal to 0.542 and 0.969, respectively). The MCC and ACC of our proposed method are also much better than those of the other sequence-based prediction methods. The results indicate that our proposed method has good generalization ability for ATP-binding sites prediction.
Case study
Inspired by previous works [Citation18, Citation19, Citation32], in order to show the prediction performance more visually, we display the prediction result of the protein sequence whose PDB id is 1O93_A in . The 1O93_A shares less than 40% sequence identity with the sequences in ATP-227; therefore, the ATP-227 dataset is used as the training set. Besides our proposed method, predictors constructed on ATP-227 with the under-sampling technique and the original ATP-227 dataset without under-sampling are utilized. At the same time, predictor TargetATP, which shows second best performance in the performance comparison section, is also involved. The result of TargetATP is obtained through its online-service (http://www.csbio.sjtu.edu.cn:8080/TargetATPsite/).
Figure 6. Binding sites predicted by the proposed method, predictor constructed on under-sampled dataset, predictor constructed on original dataset and TargetATP for protein 1O93_A.
Note: Black dots indicate the true binding sites in the protein sequence; blue dots indicate the binding residues predicted by the proposed method; red, orange and green dots represent the result of TargetATP, predictor constructed on under-sampled dataset and predictor constructed on original dataset, respectively. The probability curves of involved predictors are drawn in corresponding colors.
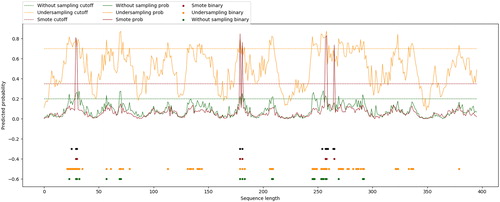
It can be observed that the predictor based on under-sampling correctly predicts 8 binding residues among 11 true binding residues, but with 61 false positive samples at the same time (). The predictor based on original dataset correctly predicts 7 binding residues with 16 false positive samples. TargetATP correctly predicts 5 binding residues with 5 false positive samples. However, our proposed method correctly predicts 7 of 11 true binding residues with no false positive samples. The MCC of predictors based on the under-sampled dataset, the original dataset, TargetATP and the proposed method on 1O93_A is 0.246, 0.418, 0.462 and 0.793, respectively. The prediction result indicates that our proposed method has better identifying ability of ATP-binding residues and efficiently reduces the number of false positive samples, which is important for further biological experiments.
Conclusions
In this study, we developed a sequence-based prediction method for ATP-binding sites in protein sequences. Compared with previous methods, our proposed method utilizes the SMOTE algorithm, which does not lose any information in the dataset to solve the imbalanced learning problem. With the combination of Random Forest classifier, which provides satisfying training speed and classification performance, our method obtains better performance than predictors trained with the under-sampled dataset or the original dataset. In order to further improve the prediction performance, a complementary template-based method which detects homology of small segments in the query sequence is also developed. The results of the two prediction methods are merged to generate the final prediction. Compared with other sequence-based prediction methods, our proposed method obtains higher MCC and AUC values over five-fold cross-validation on benchmark datasets. On the independent dataset that is used to reveal the generalization ability, our proposed method keeps the stable performance, which demonstrates that our method can reliably predict the ATP-binding sites in protein sequences and provide assistance for experimental researchers.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in BMC Bioinformatics at https://doi.org/10.1186/1471-2105-10-434, reference [Citation17], and in Proteome Science at https://doi.org/10.1186/1477-5956-9-s1-s4 [Citation18].
Additional information
Funding
References
- Ke R, Xu Q, Li C, et al. Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism. Cell Biol Int. 2018;42(4):384–392.
- Suzuki R, Hotta K, Oka K. Transitional correlation between inner-membrane potential and ATP levels of neuronal mitochondria. Sci Rep. 2018;8(1):2993–3004.
- Kojima S, Ohshima Y, Nakatsukasa H, et al. Role of ATP as a key signaling molecule mediating radiation-induced biological effects. Dose Response. 2017;15(1):1559325817690638–1559325817690611.
- Mizushima T, Nishida S, Kurokawa K, et al. Negative control of DNA replication by hydrolysis of ATP bound to DnaA protein, the initiator of chromosomal DNA replication in Escherichia coli. Embo J. 1997;16(12):3724–3730.
- Protein Data Bank. http://www.rcsb.org [cited Jul 2020].
- Hu J, Li Y, Zhang Y, et al. ATPbind: accurate protein-ATP binding site prediction by combining sequence-profiling and structure-based comparisons. J Chem Inf Model. 2018;58(2):501–510.
- Chaudhry C, Farr GW, Todd MJ, et al. Role of the gamma-phosphate of ATP in triggering protein folding by GroEL-GroES: function, structure and energetics . Embo J. 2003;22(19):4877–4887.
- Goldin N, Heyfets A, Reischer D, et al. Mitochondria-mediated ATP depletion by anti-cancer agents of the jasmonate family. J Bioenerg Biomembr. 2007;39(1):51–57.
- Pitman MR, Powell JA, Coolen C, et al. A selective ATP-competitive sphingosine kinase inhibitor demonstrates anti-cancer properties. Oncotarget. 2015;6(9):7065–7083.
- Sasaki N, Ishiwata T, Hasegawa F, et al. Stemness and anti-cancer drug resistance in ATP-binding cassette subfamily G member 2 highly expressed pancreatic cancer is induced in 3D culture conditions . Cancer Sci. 2018;109(4):1135–1146.
- Kumar S. Prediction of metal ion binding sites in proteins from amino acid sequences by using simplified amino acid alphabets and random forest model. Genomics Inform. 2017;15(4):162–169.
- Srivastava A, Kumar M. Prediction of Zinc binding sites in proteins using sequence derived information. J Biomol Struct Dyn. 2018;36(16):4413–4423.
- Gao J, Bo Z, Zhu J, et al. Predicting protein ligand binding sites with structure alignment method on Hadoop. CP. 2016;13(2):113–121.
- Jia J, Liu Z, Xiao X, et al. Identification of protein-protein binding sites by incorporating the physicochemical properties and stationary wavelet transforms into pseudo amino acid composition. J Biomol Struct Dyn. 2016;34(9):1946–1961.
- Maheshwari S, Brylinski M. Prediction of protein-protein interaction sites from weakly homologous template structures using meta-threading and machine learning. J Mol Recognit. 2015;28(1):35–48.
- SWISS-PROT Database. https://www.uniprot.org. Accessed February, 2019.
- Chauhan JS, Mishra NK, Raghava GP. Identification of ATP binding residues of a protein from its primary sequence. BMC Bioinformatics. 2009;10(1):434–442.
- Chen K, Mizianty MJ, Kurgan L. ATPsite: sequence-based prediction of ATP-binding residues. Proteome Sci. 2011;9(Suppl 1):S4. DOI: https://doi.org/10.1186/1477-5956-9-s1-s4
- Chen K, Mizianty MJ, Kurgan L. Prediction and analysis of nucleotide-binding residues using sequence and sequence-derived structural descriptors. Bioinformatics. 2012;28(3):331–341.
- Yu DJ, Hu J, Huang Y, et al. TargetATPsite: A template-free method for ATP-binding sites prediction with residue evolution image sparse representation and classifier ensemble. J Comput Chem. 2013;34(11):974–985.
- Andrews BJ, Hu J. TSC_ATP: A two-stage classifier for predicting protein-ATP binding sites from protein sequence. 2015 IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB). IEEE, 2015.
- Chawla NV, Bowyer KW, Hall LO, et al. SMOTE: synthetic minority over-sampling technique. JAIR. 2002;16(1):321–357.
- Blagus R, Lusa L. Evaluation of SMOTE for high-dimensional class-imbalanced microarray data. International Conference on Machine Learning & Applications. Miami Florida, USA, 2013.
- Jeatrakul P, Wong KW, Fung CC. Classification of imbalanced data by combining the complementary neural network and SMOTE algorithm. LNCS. 2010; 6444(Part 2):152–159.
- Bauer RA, Günther S, Jansen D, et al. SuperSite: dictionary of metabolite and drug binding sites in proteins. Nucleic Acids Res. 2009;37(Database issue):D195–D200. (Database issue):
- Fu L, Niu B, Zhu Z, et al. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28(23):3150–3152.
- Mcguffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16(4):404–405.
- Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402.
- Faraggi E, Zhou Y, Kloczkowski A. Accurate single-sequence prediction of solvent accessible surface area using local and global features. Proteins Struct Funct Bioinform. 2014;82(11):3170–3176.
- Yang X, Wang J, Sun J, et al. SNBRFinder: a sequence-based hybrid algorithm for enhanced prediction of nucleic acid-binding residues. PLOS One. 2015;10(7):e0133260. DOI: 10.1371/journal.pone.0133260.
- Walia RR, Xue LC, Wilkins K, et al. RNABindRPlus: a predictor that combines machine learning and sequence homology-based methods to improve the reliability of predicted RNA-binding residues in proteins. PLOS One. 2014;9(5):e97725. DOI: 10.1371/journal.pone.0097725.
- Yu DJ, Hu J, Tang ZM, et al. Improving protein-ATP binding residues prediction by boosting SVMs with random under-sampling. Neurocomputing. 2013;104:180–190.