
Abstract
The formation of adventitious roots (ARs) plays an important role in grass growth and production. Chicken alpha interferon (ChIFNα) is an animal glycoprotein with broad activities of immune control, gene induction, growth and development regulation. ChIFNα gene was transformed to Lotus japonicus and its expression was found to promote AR development. The primordia formation occurred 3–5 days earlier, and more root hairs appeared in the transgenic plants (TP) than in the wild-type parents (WT). Data of RNA sequences from TP and WT in vitro hydroponic culture were analyzed. Overall, the intersection of the differentially upregulated genes at 0–12 days included 278 genes in TP and 244 genes in WT plants. Gene Ontology analysis indicated that the differentially expressed genes (DEGs) in TPs were enriched in response to biotic stimulus, metabolic process, sucrose transport, carbon fixation and signal transduction. Further, Kyoto Encyclopedia of Genes and Genomes enrichment analysis found carbon metabolism, carbon fixation in photosynthetic organisms and photosynthesis enriched in TPs. Co-expression analysis of the DEGs showed that 35 and 18 transcription factors (TFs) were involved in ARs formation in TP and WT plants, respectively. However, the TFs C3H and C2C2-GATA showed the opposite regulatory pattern with ChIFNα expression. Together, these data suggest that ChIFNα promotes AR development by modulating specific pathways and TFs. These results provide new insight into the mechanism by which animal interferon modulates plant growth and lays the foundation for cultivating new forage germplasm using animal cytokines.
Introduction
Lotus japonicus L. is a perennial herb of the Leguminosae family, and is distributed in southwest and northwest China. It is not only a high-quality perennial leguminous forage plant and honey source, but is also important for water and soil conservation due to its developed root system and strong root propagation abilities [Citation1]. L. japonicus has many beneficial characteristics, such as a strong capacity for nitrogen fixation, tender branches and leaves, high nutritional value, high protein content, good palatability, strong grazing resistance, and easy digestion by domestic animals [Citation2]. At the same time, the creeping roots of the lotus can form a developed network for soil retention, and reduce the loss of nutrients, such as nitrogen, phosphorus, and potassium, which make it a good candidate for applications in soil and water conservation and soil improvement. Further, it shows a high flowering rate, a high seed setting rate, and stable inheritance of these many beneficial traits [Citation3]. These features make it amenable to genetic engineering. In addition, it is easy to introduce foreign genes into the lotus, as it exhibits a relatively high transformation efficiency, and the transgenic plants (TPs) are easy to grow. Therefore, the lotus plays an important role in plant genetic engineering and nitrogen fixation engineering, and is an ideal material for forage quality optimization and animal vaccine production in bioreactors [Citation4].
Interferons (IFN) are a group of multifunctional animal cytokines, which are small, soluble polypeptides produced in response to exogenous stimuli with a range of biological functions, including hormone-like effects, immune regulation, antiviral functions, and stress reduction, among others [Citation5–7]. Studies have shown that IFN produced by humans and other animals, as well as the IFN-stimulating 2-5-oligoadenylate, can be transformed into plants and influence their behavior [Citation8–10]. In addition, safe and reliable biologically-active protein can be produced by transforming plants with IFN, which can then be used in medical treatments [Citation11,Citation12]. The HuIFN-α/β and chicken interferon genes (ChIFN) have been successfully expressed in Cucumis sativus (cucumber), Cucurbita moschata (pumpkin), Lactuca sativa (lettuce), Oryza sativa (rice), Nicotiana tabacum (tobacco), and other plants, and the recombinant interferon proteins (rIFNs) show physiological functions similar to those in animal cells [Citation13–17]. Chicken alpha interferon (ChIFN-α) was the first IFN to be discovered and thoroughly studied, and its coding sequence was first obtained in 1994 [Citation18]. Studies have shown that ChIFN-α can promote growth and development, as well as inhibit virus proliferation, in both animals and plants [Citation19,Citation20]. However, the mechanism by which it regulates plant development remains unclear. Although there are many distinctions between animals and plants, they share similar regulatory mechanisms. For example, melatonin, which is involved in the regulation of circadian rhythms, growth and development, and immunity in animals, is also found in plant cells [Citation21]. In addition, soybean flavone has estrogen-like effects in animals. Likewise, although animal IFN has been shown to exert multiple effects in plants, the detailed molecular mechanisms regulating these processes are still unknown.
Root systems are an important nutrient organ that formed during the evolution of plants. They are typically composed of a primary root and lateral roots (LRs), but may also include ARs [Citation22,Citation23]. These act as channels for material exchange between plants and soil, and play an important role in absorbing nutrients and water during plant growth. ARs are uniquely formed from tissues other than roots, including tender stems, branches, overground stems, and underground stems, among others [Citation24]. Adventitious rooting is the most important mechanism underlying vegetative propagation and an important strategy for plant propagation under environmental stress [Citation25]. To experimentally study ARs, cuttage and hydroponic hormone induction are the most common methods used to form ARs, and their growth involves the sequential steps of: a) dedifferentiation; b) cell division; and c) adventitious root primordia initiation, development, and outgrowth [Citation26–28]. ARs have a series of special biological functions, as they only form under certain conditions and in certain positions. These features directly lead to the difficulty of plant rooting, and are the limiting factors for forage yield and rapid grassland formation. Therefore, research on ARs has important theoretical and practical significance for understanding the growth and development of plants, and the vegetative propagation of plants.
Transcriptome sequencing has become a means of quickly revealing the key genes, or related gene networks, responsible for root formation. For example, transcriptome technology was used to analyze the molecular regulatory network of root development in Chinese fir and Panax ginseng [Citation29,Citation30]. Similarly, this method was used to reveal the Gene Ontology (GO) of genes involved in the growth and development of ARs, which include cell carbohydrates, photosystems, polysaccharide metabolism, carbohydrate metabolism, and cell carbohydrate metabolism [Citation26,Citation31,Citation32]. Additional genes related to AR formation, including photosynthesis and the starch and sucrose metabolism pathways, were revealed by comparison with the Kyoto Encyclopedia of Genes and Genomes (KEGG) database [Citation31,Citation33].
In this study, the animal interferon gene ChIFNα was transformed into the herbage model plant L. japonicus in order to better understand its bioactivity and regulatory mechanisms. Using microscopic observation, RNA-Seq and qRT-PCR experiments, we examined the developing ARs in the transgenic ChIFNα L. japonicus (TPs) and WT plants. We found that TP ARs grew 3–5 days earlier than those in WT plants. GO and KEGG enrichment analysis showed that the differentially expressed genes (DEGs) in TP were enriched in the terms: response to biotic stimulus, metabolic process, sucrose transport, carbon fixation, and signal transduction, including carbon metabolism, carbon fixation in photosynthetic organisms, and photosynthesis. These results suggest that ChIFNα mediates AR developmental regulation by modulating specific pathways and TFs. Co-expression analysis of DEGs showed that 35 and 18 transcription factors (TFs) were involved in AR formation in TP and WT plants, respectively. Our study reveals that the animal cytokine ChIFNα may promote plant AR development through the upregulation of metabolism-related genes and molecular networks mediated by root regulatory TFs. This study provides a theoretical basis for grass growth regulation, molecular breeding, and the rooting practice of plant cutting.
Materials and methods
Transformation and identification of transgenic plant
ChIFNα gene (GenBank no. U07868) was introduced into L. japonicus plant by Agrobacterium-mediated transformation method using the stem as explant. ChIFNα gene and GUS reporter gene were driven by CaMV 35S promoter in the transformed plasmid pSFRLH [Citation34]. Expression of GUS gene in leaf tissues of transgenic plants (TP) was examined by histochemical assay according to Song et al. [Citation35]. Plant DNA extraction kit (DP305) from Tiangen (Beijing, China) was used to extract the DNA of GUS positive plants and wild-type plants (WT). The ChIFNα target gene was detected by polymerase chain reaction (PCR) using primers P1 5′-GCTGTTCCAGCTTCTCCAC-3′ and P2 5′-CCTGGTGTTTCCGGTAAGG-3′, which amplified the target fragment of 604 bp. The PCR reaction included 1 μL DNA, 12.5 μL 2× Taq plus PCR master mix (Aidlab, Beijing, China), 0.5 μL each of forward and reverse primers, and 25 μL ddH2O. The PCR procedure was performed using the C1000TM Thermal Cycler (Bio-Rad, CA, USA) under the following conditions: 94 °C for 5 min, 30 cycles of 94 °C for 40 s, 56 °C for 30 s, 72 °C for 30 s, and a final extension of 8 min at 72 °C.
Transcriptome plant materials
The genome of L. japonicus is derived from (V3.0) (http://www.kazusa.or.jp/lotus/). Two lines (TP and WT) of L. japonicus were grown in the greenhouse of the Institute of Agricultural Biotechnology of Guizhou University (Guiyang, Guizhou, China, 106° 675′ E; 26° 427′ N). Branches from TP and WT with the same extent of growth were isolated, and their stem bases were cut into three groups, ensuring that each cut stem was a branch with leaves. These were then fixed with a floating plate, placed in tap water for hydroponics, and cultured in a light incubator at 20 °C. The light cycle for the culture was set as 16 h of light and 8 h of dark, and the light intensity was 15,000 LX, which was changed every 1 day. 10 Samples from TP and WT were taken at 0 days after hydroponics (DAH), 3 DAH, 6 DAH, 9 DAH, and 12 DAH. Samples were prepared by mixing 120–150 lines from different periods, and then stored in liquid nitrogen immediately after sampling.
Microscopic observation of ARS
Using sterilized tweezers, the hydroponically developed ARs were gently removed, cut into about 0.1–0.2 mm pieces, soaked and fixed with 2.5% glutaraldehyde solution, and placed in a refrigerator at 4 °C overnight. The samples were then washed 3 times with 0.1 mol/L (pH = 7.2) phosphate buffer solution for 10 min each. They were then dehydrated in 55%, 65%, 75%, 85%, 95%, and 100% ethanol solutions (with sterile water) for 10 min each at room temperature. The excess ethanol remaining on the dehydrated glass slide was drained off and the slides were freeze dried for 60–120 min. The samples were then observed under a microscope. Samples with well-preserved morphology were selected, adhered to the sample table with conductive adhesive, and gold films were sprayed on using an ion sputtering apparatus. After coating, the samples were observed using a scanning electron microscope Hitachi S-3400N.
Total RNA extraction and transcriptome sequencing
RNA extraction from the lotus roots was carried out using an RNApre Pure plant RNA extraction kit (DP432) from Tiangen (Beijing, China). RNA concentration was measured with a NanoPhotometer NP80 Touch spectrophotometer (IMPLEN, Germany), and RNA integrity was confirmed by assessing that the samples had an RNA integrity number (RIN) of 1.8–1.9. RNA solutions for the 10 samples were stored at −80 °C. A total of 1–2 µg RNA per sample with 28S/18S RNA ratio ≥ 1.8 were used for library preparation. The mRNA sequencing library was constructed using the NEBNext® Ultra RNA Library Prep Kit (New England Biolabs Inc., Ipswich, MA, USA). The sequencing library was analyzed using the Agilent 2100 Bioanalyzer with a minimum integrity number (RIN) value of 10 and the insertion elements were 250–300 bp. The library was sequenced using an Illumina HiSeq™ 2000 sequencing system (Illumina Inc., San Diego, CA, USA). The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive in National Genomics Data Center, Beijing Institute of Genomics (China National Center for Bioinformation), Chinese Academy of Sciences, under accession number CRA002426 that are publicly accessible at https://bigd.big.ac.cn/gsa [Citation36,Citation37].
Transcriptome analysis
Transcriptome data analysis was divided into the following steps. First, QoRTs software was used to filter the 10 groups of data, producing ‘clean’ data [Citation38]; at the same time, it was beneficial to carry out quality control of the data, and select data that met the standard for subsequent analysis. Second, STAR v2.7.0 was used to construct genome index files and comparisons [Citation39]. Third, HTSeq-count was used to quantify transcript expression [Citation40]. Finally, DESeq2 was used to analyze the differences between the different groups [Citation41]. The DEG numbers were compared between TP and WT at 0, 3, 6, 9 and 12 days. The set of genes from those DEGs obtained in the first step from each stage of TP or WT were then counted. The genes in the two sets (TP and WT groups) were considered to be DEGs. FPKM values (fragments per kilobase of exon per million fragments mapped) were determined from the abundance of standardized genes. The absolute value of |log2Foldchange| >1 and the false discovery rate < 0.05 was used to identify the significance of the changes in the expression levels of different genes. Subsequently, Clusterprofiler was used to enrich and analyze the DEGs using the GO and KEGG databases[Citation42] .
Analysis of gene co-expression network and quantitative RT-PCR analysis
Python scripts were used to analyze the FPKM data for the transcriptomes of TP and WT L. japonicus plants, and DEGs and TFs were used to determine the co-expression network [Citation43]. Comments on the TFs of L. japonicus are from the database iTAK [Citation44]. The co-expression results were visually analyzed using Cytoscape v3.7.10 [Citation45]. Quantitative RT-PCR was used to detect the expression levels of genes from the transcriptome data of 10 groups of L. japonicus. First strand cDNA synthesis was performed using the first strand cDNA synthesis kit from Thermoscientific Aid First Strand. The reaction consisted of 2 μL total RNA, 1 μL oligo (dT)18 primer, and ddH2O to 12 μL. The reactions were centrifuged at low speed for 5 min at 65 °C, cooled on ice, centrifuged at low speed, and cooled again. To the reactions were added 4 μL X Reaction Buffer, 1 μL RiboLockTM RNA inhibitor (20 U/μL), 2 μL 10 mmol/L dNTP Mix, 1 μL RevertAidTM M-MuLV reverse transcriptase (200 U/μL), and ddH2O to 20 μL. They were then centrifuged at low speed and mixed, and incubated at 42 °C for 60 min and then 70 °C for 5 min.
Ubiquitin and actin were used as reference housekeeping genes for L. japonicus. Fluorescent quantitative gene nucleic acid sequences were designed with fluorescent primers (Supplemental Table S1) using Primer3plus software [Citation46]. Ten samples were used for cDNA fluorescence quantitative analysis using a 96 Real-time qTOWER3.0 (Analytikjena, Germany). The PCR reaction included 10 μL SYBR Primix EX Taq TM II (Zombio, Beijing, China), 1 μL each of forward and reverse primers, and ddH2O to 20 μL. The PCR reaction procedure was 95 °C for 30 s; 95 °C for 15 s; 60 °C for 30 s; and 72 °C for 1 min, for 40 cycles. Quantitative RT-PCR results were analyzed using the 2–△△Ct method [Citation47]. Heat maps were drawn using TBtools [Citation48].
Results and discussion
Identification of transgenic plants
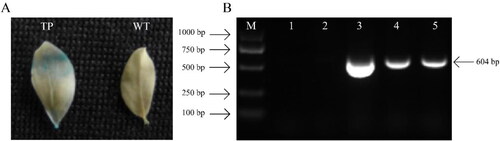
Transgenic L. japonicus was obtained for positive GUS histochemical staining and PCR reaction. Obvious GUS activity was detected in the leaves transformed with ChIFNα gene and evident blue inclusions were observed (). PCR assay yielded a positive result and 604-bp amplification bands were found in the transformed line but not in the wild type (). The results indicated that ChIFNα foreign gene had been successfully transferred to the L. japonicus grass.
Figure 1. Molecular analysis of transgenic L. japonicus plants. GUS assay (A) of transformed plant; TP: transgenic plants; WT: wild type; blue stain exhibited GUS activity in TP leaf. PCR analysis (B) of transgenic plant; M: DL2000 Marker (TaKaRa, Dalian, China); 1: H2O; 2: Wild type; 3: pSFRLH plasmid carrying ChIFNα gene; 4–5: Different transformed lines using ChIFNα gene.
Microscopic observation of hydroponically-grown ARs
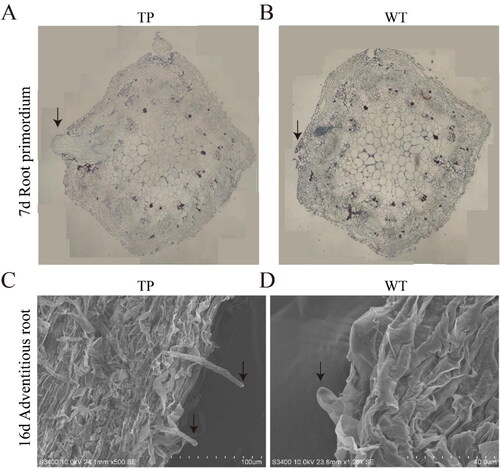
In order to compare the root development differences between TP and WT lines, the root stem nodes, which correspond to the sites of AR formation, from L. japonicus grown for 7 and 16 days in the in vitro hydroponic system were prepared as paraffin sections and then imaged using a scanning electron microscope (). These results indicated that TP developed more efficiently than WT L. japonicus. At the point at which the TPs had formed root primordia, the WT plants had just germinated (). The root primordia of TP ARs were formed 3–5 days earlier than those in WT plants. In addition, the roots were longer and more numerous in TPs compared to WT plants. To examine the differences in the development of the stem tips at later stages of hydroponic growth, we compared root hairs from TPs and WT plants on the 16th day of growth using electron microscopy. The root hairs of TPs were longer and more numerous than those in WT plants on the same day of growth (). Taken together, these results indicate that the ARs in TPs developed earlier than those in WT plants. The growth of ARs, root primordial, and root hairs in TPs were better developed than those in WT plants under the same growth period, indicating that the ChIFNα gene promoted the development of L. japonicus ARs. This suggests that ChIFNα could also more generally promote root development in L. japonicus.
Figure 2. Paraffin sections and electron microscopy of ARs from TP and WT L. japonicus. (A,B) Radical paraffin sections at 7 DAH of TP (A) and WT (B); the arrow points to the root primordium; (C,D) Electron micrograph at 16 DAH of TP (C) and WT (D); the arrow points to the indefinite root.
The animal IFN genes ChIFN-α and ChIFN-γ, when expressed in plants, can affect resistant growth and glandular hair density [Citation19,Citation49,Citation50]. In the present study, ChIFN-α gene expression in L. japonicus affected AR formation and development. During plant growth, auxin plays an important role in regulating root development, which is closely related to the development of ARs [Citation51]. Ethylene, indoleacetic acid, and methyl jasmonate are also known to participate in the development of LRs and ARs. Continuous developments in biotechnology have allowed hormones to be used to improve plant growth. However, the extensive use of hormones in plants poses potential risks to animal health and human food safety [Citation52]. It is therefore important to also study non-hormonal regulators of herbage plants, like ChIFNα. Our study found that ChIFNα is similar to plant hormones in its ability to regulate growth. These results improve our understanding of the role played by animal interferons in plants and suggest how they be used more efficiently in plant production.
Transcriptome sequencing
cDNA from 10 TP or WT plant samples were sequenced using the Illumina sequencing platform. Each sample generated more than 6 Gb of data and a total of about 60 Gb of high-quality data was obtained. As shown in (Supplemental Table S2), the raw data for each of the samples were all over 40 million reads. After filtering out the invalid data, the clean reads showed few changes compared with the raw reads, with a GC content of about 43%, a Q20 ratio of over 95%, and a Q30 ratio of over 90%, indicating that the sequencing results were valid.
Table 1. Statistics on sequencing data of two different varieties of lotus root transcriptome.
Differentially expressed genes (DEGs)
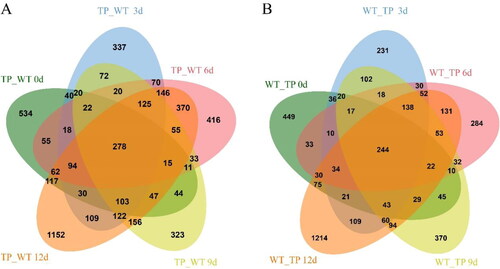
During the development of ARs at 0 d, 3 d, 6 d, 9 d and 12 d, we found that 1490, 1606, 1790, 1446 and 2981 genes, respectively, were up-regulated in TPs. Over the same developmental period in WT plants, there were 1118, 1165, 1138, 1297 and 2349 up-regulated genes, respectively. This indicates that there were fewer genes transcribed in WT plants than in TPs during the same developmental stages, suggesting that the early root development observed in TPs may be attributed to the increased expression of genes regulating root development (). Next, the DEGs in TPs and WT plants at different root development stages (0d, 3d, 6d, 9d and 12d) were analyzed. We found that the common up-regulated DEGs were 278 and 244 in the TP group (0–12 days) and WT group (0–12 days). Thirty-four additional genes were up-regulated in TPs than in the WT plants with delayed root development (Supplemental Table S3). Thus, the TP transcriptome analysis showed that the number of DEGs increased significantly compared to WT plants. This suggests that the difference in AR development between TPs and WT plants was related to differences in developmental regulation. We speculated that the ChIFNα gene could regulate the development of ARs through a molecular regulatory network.
Figure 3. DEGs Venn diagram of L. japonicus. (A) The intersection of up-regulated genes in TPs and WT plants at different growth times; (B) The intersection of up-regulated genes in WT plants, representing differences between WT and TP at different growth times.
Enrichment analysis of DEGs
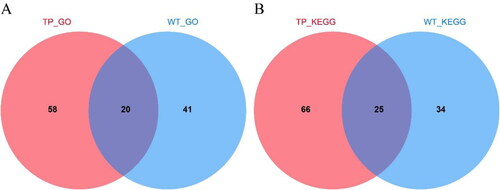
Based on the results of the DEGs analysis, we next performed an enrichment analysis on the DEGs in the different groups (Supplemental Tables S4 and S5). GO and KEGG enrichment analysis of the DEGs in TPs and WT plants indicated that TPs and WT plants had 20 shared processes in GO enrichment; 58 processes only in TP and 41 processes only in WT. There were 25 shared pathways in KEGG enrichment, while 66 pathways only in TP and 34 pathways only in WT ().
Figure 4. Venn diagram of TP and WT enrichment analysis differences. (A) Venn diagram of TP and WT DEGs GO enrichment analysis; (B) Venn diagram of KEGG enrichment analysis of TP and WT DEGs.
In GO enrichment analysis, there were 277 genes in TP (excluding ChIFNα), and a total of 213 were annotated. There were 69 genes belonging to ‘biological processes,’ including biological processes (17, 7.98%), transport (9, 4.22%), biosynthetic process (5, 2.35%) and transmembrane transport (5, 2.35%). There were 45 genes belonging to ‘cellular component,’ including cellular component (16, 7.51%), nucleus (6, 2.82%) and protein complex (4, 1.88%). There were 99 genes belonging to ‘molecular functions,’ including molecular functions (27, 12.68%), ion binding (15, 7.04%) and DNA binding (10, 4.70%). However, 164 genes were annotated from 244 genes in WT. There were 52 genes belonging to ‘biological process,’ including biological process (8, 4.88%), cellular protein modification process (5, 3.05%), biosynthetic process (4, 2.44%), lipid metabolism process (4, 2.44%) and stress response. There were 33 genes belonging to ‘cellular component,’ including cellular component (15, 9.15%), nucleus (6, 3.66%), and Golgi apparatus (2, 1.22%). There were 79 genes belonging to ‘molecular functions,’ including molecular functions (24, 14.63%), ion binding (18, 10.99%) and kinase activity (5, 3.04%). At the same time, the intersection of TP and WT in the DEGs enrichment set included defense reaction (GO:0006952, 1:3), response to biological stimulation (GO:0009607, 1:1), transporter activity (GO:0005215, 2:1) and translation (GO:0006412, 2:1). TP contained sucrose transport (GO:0015770, 1, p = 0.032), sucrose transmembrane transport activity (GO:0008515, 1, p = 0.035), transcription factor binding transcription factor activity (GO:0000989, 1, p = 0.035), and carbon fixation (GO:0015977, 1, p = 0.038) ().
Figure 5. Enrichment analysis of DEGs GO. TP (A) and WT (B) GO enrichment analysis.
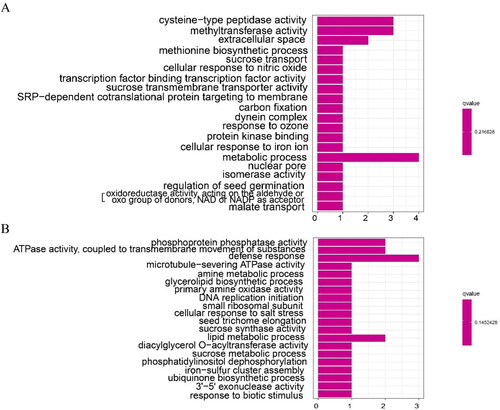
The GO analysis of the transcriptome data showed that there were 17 more genes in the TP GO enrichment process than for WT plants. TP and WT shared other processes, such as biological stimulation, RNA binding, transportation, translation and metabolism. The transport, carbohydrate metabolism, sucrose transport, sucrose transmembrane transporter activity, carbon fixation, flavonoid biosynthesis, carbohydrate binding, signal transduction and other processes were differentially expressed only in TPs. Studies on ARs from Cryptomeria fortunei showed that the GO enrichment mainly concentrates on carbohydrate, phytohormone and other processes, and it also contained signal transduction defense reaction [Citation33]. Similarly, the cellular functions of the AR transcriptome in mulberry included RNA transport and sucrose metabolism [Citation32]. These regulatory pathways, including transportation, are closely related to root development [Citation26]. The results of the GO enrichment pathway analysis thus suggest that the better development of ARs in TPs was likely due to changes in pathways related to root growth and development.
In KEGG enrichment analysis, a total of 277 genes in TP (excluding ChIFNα) were enriched in 49 KEGG pathways, including cellular processes (4, 8.16%), environmental information processing (6, 12.24%), genetic information processing (19, 38.78%), metabolism (32, 65.31%) and organismal systems (13, 26.53%). However, only 38 KEGG pathways were enriched from 244 differential genes in WTs, including Cellular Processes (10, 26.32%), Environmental Information Processing (12, 31.58%), Genetic Information Processing (13, 34.21%), Metabolism (16, 42.11%) and Organismal Systems (14, 36.84%). The KEGG enrichment set for the TP and WT intersection included biosynthesis of secondary metabolites (lja01110, 5:3), Metabolic pathways (lja01100, 11:3), and DNA replication (lja03030, 1:3). TP only included Carbon metabolism (lja01200, 3), Phenylpropanoid biosynthesis (lja00940, 2), Pyruvate metabolism (lja00620, 2), Citrate cycle (TCA cycle) (lja00020, 2), Carbon fixation in photosynthetic organisms (lja00710, 1), RNA transport (lja03013, 2), and Photosynthesis (lja00195, 1), among others (Supplemental Table S4; ).
Figure 6. Enrichment analysis of DEGs KEGG. TP (A) and WT (B) KEGG enrichment analysis.
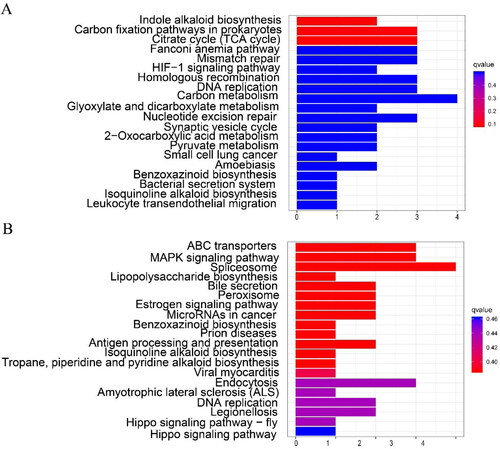
The KEGG enrichment analysis for TPs mainly revealed flavonoid biosynthesis, carbon metabolism, TCA cycle, fixation in photosynthetic organisms, and photosynthesis. During mulberry root development, hormone signal transduction, energy metabolism and starch metabolism, photosynthesis, sugar metabolism, and other processes were all found to be enriched [Citation53]. Polysaccharide metabolism, carbohydrate metabolism, and photosynthesis were also enriched during ramie AR formation [Citation31]. Taken together, these results suggest that the development of TP ARs is regulated by the above enriched processes and pathways, which are known to be involved in plant root growth, and involves more differentially expressed genes than WT.
Analysis of co-expression networks of DEGs and TFs
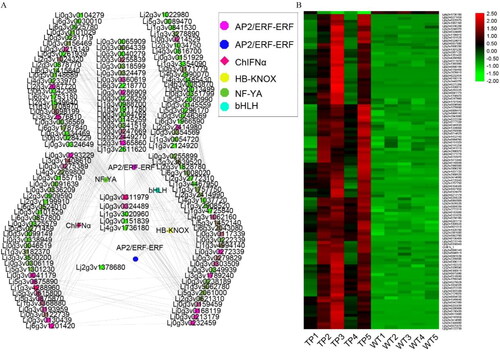
Co-expression analysis of the 278 and 244 DEGs in the transcriptomes of TP and WT, respectively, showed that the correlation coefficient (R) was greater than 0.97. Overall, 102 genes were co-expressed, with 35 TFs in TP, and 105 genes were co-expressed, with 18 TFs in WT (Supplemental Tables S6, S7 and S8; ). There were 5 TFs in TP and 2 TFs in WT that were different in the DEGs. The expression levels of the TFs in TPs were significantly higher than those in WT. In TP, the top four TFs were C3H (Lj0g3v0308719, degree = 20), C2C2-GATA (Lj3g3v3386580, degree = 15), GRAS (Lj0g3v0182089, degree = 13), and HB-other (Lj2g3v1863470, degree = 13); C3H and C2C2-GATA were negatively correlated TFs, while GRAS and HB-other were positively correlated TFs. In addition, co-expressed TFs with degree ≥ 3 in the co-expression analysis included NAC (Lj0g3v0223189), AP2/ERF-ERF (Lj1g3v1676620), bZIP (Lj5g3v1949500), FAR1 (Lj1g3v3556900), AP2/ERF-ERF (Lj6g3v2007160), bHLH (Lj5g3v2030840), C3H (Lj0g3v0180079), and zf-HD (Lj1g3v0244390), among which C3H (Lj0g3v0180079) was positively correlated in co-expression, while bHLH (Lj5g3v2030840) was negatively correlated ().
Figure 7. Network diagram of the co-expression of DEGs and TFs. DEGs co-expressed with TFs in TP (A) and WT (B); the solid line indicates positive correlation and the dashed line indicates negative correlation.
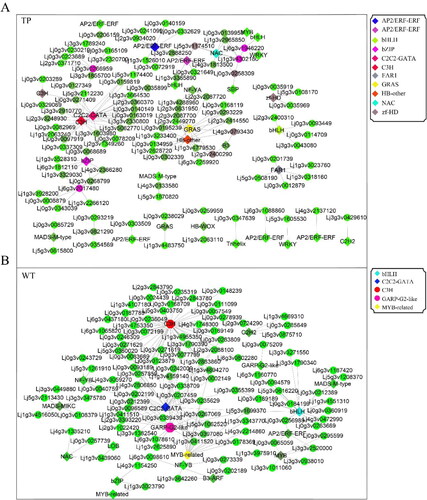
In WT, the top five TFs were C3H (Lj0g3v0308719, degree = 41), C2C2-GATA (Lj3g3v3386580, degree = 36), GARP-G2-like (Lj3g3v3386580, degree = 17), bHLH (Lj3g3v3033250, degree = 15), and MYB-related (Lj6g3v0933490, degree = 13). Among them, all except C2C2-GATA were negatively correlated, while the other four TFs were positively correlated. In addition, TFs with degree ≥ 3 in the co-expression analysis included MYB (Lj0g3v0165839, degree = 6), MADS-MIKC (Lj5g3v2029620, degree = 5), B3-ARF (Lj1g3v2155550, degree = 4), AP2/ERF-ERF (Lj1g3v4241090, degree = 4), C2H2 (Lj4g3v1719700, degree = 4), GARP-G2-like (Lj0g3v0195339, degree = 3), NAC (Lj1g3v4579400, degree = 3), and NF-YB (Lj6g3v1584620, degree = 3). Among them, MYB, AP2/ERF-ERF, and C2H2 were negatively correlated in co-expression, while MADS-MIKC, B3-ARF, GARP-G2-like, NAC, and NF-YB were positively correlated ().
We note that C3H (Lj0g3v0308719) and C2C2-GATA (Lj3g3v3386580) were both present in the co-expression analysis of TP and WT. C2C2-GATA was positively correlated in TP co-expression and negatively correlated in WT, while C3H was negatively correlated in TP co-expression and positively correlated in WT. Therefore, we speculate that C3H and C2C2-GATA play an important role in regulating the root development in lotus, and they present opposite expression patterns in TP and WT. At the same time, the results of the co-expression and DEGs analyses showed that there were 6 DEGs in TP and 2 DEGs in WT that were TFs, which indicated that there were many TFs involved in the formation of ARs in TP that were transcriptionally regulated.
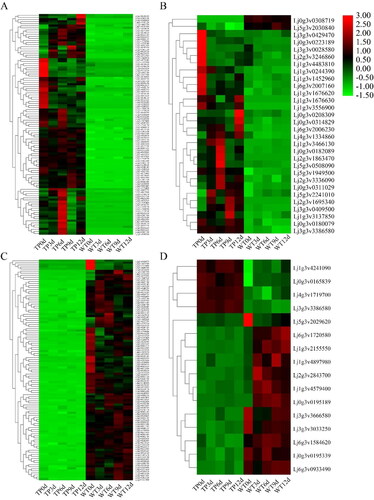
According to the heat maps (), 102 genes in TP and 105 genes in WT were expressed uniformly, and high expression existed in the corresponding 5 DAH periods of transgenic type and wild type. It can be seen from the co-expressed TFs that the same high expression level existed in each period ().
Figure 8. Heat maps of DEGs co-expressed with TFs in TP (A,B) and WT (C,D). (A,B) Heat map of 99 DEGs co-expressed with TFs (A) and 36 co-expressed TFs (B) in TP; (C,D) heat map of 105 DEGs co-expressed with TFs (C) and 18 co-expressed TFs (D) in WT; red indicates upregulation and green indicates down-regulation.
We also examined genes, and specifically TFs, that were co-expressed with ChIFNα. The results showed that 177 genes were co-expressed with ChIFNα (correlation coefficient R ≥ 0.9), of which 5 were TFs, namely bHLH, HB-KNOX, NF-YA and AP2/ERF-ERF (2 TFs). At the same time, the expression of these genes was significantly higher in TP (). These TFs have been reported to play important regulatory roles in root development. bHLH regulates the development of rice root hairs and cell formation, and at the same time regulates the growth of plant petals [Citation54,Citation55]. HB-KNOX regulates the growth and development of rhizomes [Citation56]. The expression of NF-YA can affect embryogenesis, seed morphology, and seed germination, and its family members also regulate the development of the primary and LRs of plants, plant growth, plant development, and the stress response [Citation57–59]. AP2/ERF-ERF TFs are involved in the regulation of plant growth, development, and the stress response, and also play a regulatory role in flower formation [Citation60,Citation61]. Our study reveals that the above TFs play a key regulatory role in the development of ARs upon transformation of L. japonicus with ChIFNα. These TFs may promote AR formation through multiple regulatory networks in TPs.
Figure 9. ChIFNα co-expression network and expression patterns of TFs. (A) TP co-expression network, with degree indicated; (B) co-expression network heat map.
Quantitative real-time PCR analysis
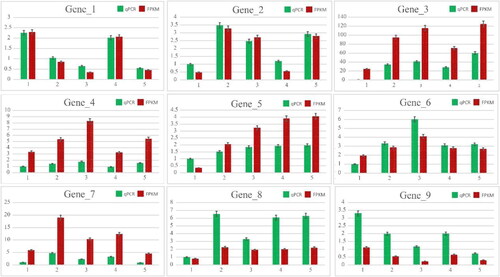
According to the transcriptome data and GO and KEGG enrichment analyses, 9 genes that were co-expressed with TFs and related to root development were selected for quantitative PCR analysis (). We examined the expression of these genes in TPs and WT plants at multiple developmental stages. Differential upregulation was found in both plant varieties, with Gene1 (Lj6g3v2007160), Gene2 (Lj3g3v3386580), Gene4 (Lj0g3v0127349) being the most obvious in the 6 DAH of WT plants, and Gene2 (Lj3g3v3386580), Gene4 (Lj0g3v0127349), Gene6 (Lj5g3v1949500) being the most obvious in the 12 DAH. Gene1 (Lj6g3v2007160), Gene2 (LjN23V3386580), Gene5 (Lj6g3v2017480) were the most obvious in the 0 DAH and Gene2 and Gene4 were the most obvious in the 6 DAH. The expression level of Gene7 (Lj0g3v0209949) was the highest at 2 DAH, but was continuously high at 3, 4, and 5 DAH. Gene 8 (Lj3g3v2320700) was expressed consistently and showed high expression at all other stages except 0 DAH. Gene3 (Lj4g3v1334860) was highly expressed in TP. The expression of Gene 9 (Lj6g3v0389080) was the highest at 0 DAH, and its expression decreased gradually over time, which may play a negative regulatory role in TP. The expression of the same genes in TPs was generally higher than in WT plants. This suggests that the formation and development of ARs in TPs was improved compared to WT plants due to the higher gene expression. Based on the electron microscopy results, the ARs in TPs exhibited improved growth and development compared to those in WT plants. In summary, 9 genes were up-regulated in TPs compared to WT plants, which was consistent with the transcriptome data.
Figure 10. qRT-PCR analysis of selected genes from transcriptomics.
Model for how ChIFNα regulates ARs development
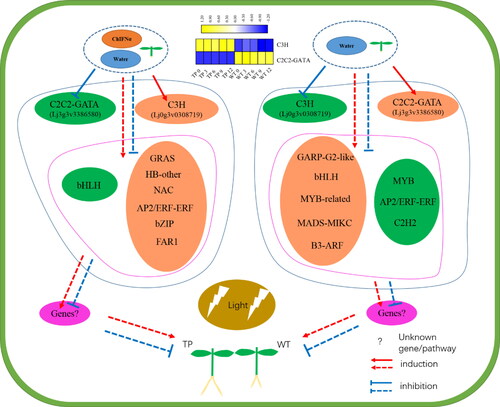
According to the co-expression results for DEGs in TP and WT, we mapped the regulatory network of ARs formation in L. japonicus (). TP roots formed 2–3 days earlier than WT roots under the same culture conditions, which may be related to the differential expression of TFs involved in the regulation of development between the two plant varieties. TFs such as C2C2-GATA (Lj3g3v3386580), GRAS (Lj0g3v0182089), HB-other (Lj2g3v1863470), NAC (Lj0g3v0223189), AP2/ERF-ERF (Lj1g3v1676620), bZIP (Lj5g3v1949500), and FAR1 (Lj1g3v3556900) were up-regulated, and TFs such as C3H (Lj0g3v0308719) and bHLH1 (Lj5g3v2030840) were down-regulated in TP plants. In WT plants, TFs such as C3H (Lj0g3v0308719), GARP-G2-like (Lj1g3v4317480), bHLH2 (Lj3g3v3033250), MYB-related (Lj6g3v0933490), MADS-MIKC (Lj5g3v2029620), and B3-ARF (Lj1g3v2155550) were up-regulated and TFs such as C2C2-GATA (Lj3g3v3386580), MYB (Lj0g3v0165839), AP2/ERF-ERF (Lj1g3v4241090), and C2H2 (Lj4g3v1719700) were down-regulated. These results show that there were marked differences in the types of TFs that were differentially expressed in the two plant varieties during hydroponic growth. Pathways that positively regulate root development were enriched in TPs compared to WT, and negative regulatory pathways were suppressed. The expression of the heterologous ChIFNα gene affected the mode by which TFs regulated development. C2C2-GATA, GRAS, HB-other, AP2/ERF-ERF, bZIP, FAR1, C3H, NAC, bHLH, GARP-G2-like, MYB, MADS-MIKC, B3-ARF, and other TFs have previously been shown to influence plant development, including reducing plant height and tillers [Citation62], plant growth and photosynthetic capacity [Citation63], root stem cell niche maintenance and root growth [Citation64], inducing early seed germination [Citation65], plant architecture [Citation66], secondary wall biosynthesis [Citation67,Citation68], root meristem development [Citation69], root and shoot development, hormonal transport, and signaling [Citation70], late stamen development [Citation71], lateral root development [Citation72] and seed and fruit development [Citation73,Citation74]. In this study, we found that ChIFNα expression induced the upregulation of the photo-sensitive TF FAR1 and HB, which participated in the regulation of root development. It is worth noting that C3H and C2C2-GATA, which were up-regulated and down-regulated, respectively, in WT plants, showed opposite regulatory processes in TPs. C3H and C2C2-GATA belong to the Zinc finger motif-containing (Znfs) family of proteins, which play a critical role in cell functions, including transcriptional regulation, mRNA degradation, and protein–protein interactions [Citation75,Citation76]. We suggest that the heterologous gene ChIFNα may function like a spanner in the lotus plant, opening or closing different regulatory paths as described above in the cells of TPs.
Figure 11. TFs and ChIFNα modulate the developmental network of ARs in L. japonicas.
Conclusions
We examined the influence of heterologous expression of ChIFNα on the development of ARs in L. japonicus grown in an in vitro hydroponic system using microscopy, RNA-Seq, and qRT-PCR. We show that TP ARs exhibited more rapid and improved development than those in WT plants, with earlier root primordia formation and more root hairs. This suggests that ChIFNα expression promoted the development of ARs in L. japonicus. According to the RNA-Seq results, more DEGs were found in TPs than in WT plants. GO and KEGG enrichment indicated that the number of DEGs and pathways differed between the two varieties of plants. Co-expression analysis of the DEGs showed that 35 and 18 TFs were involved in ARs formation in TPs and WT plants, respectively. The TFs C3H and C2C2-GATA showed opposite regulatory functions in response to ChIFNα expression. This study reveals that ChIFNα is involved in the regulation of ARs development in L. japonicus, potentially by modulating developmental regulation processes through a variety of molecular networks consisting of signaling pathways and TFs. These findings improve our understanding of how IFN influences developmental regulation in plants and supports the use of this protein in plant genetic engineering.
Supplemental Material
Download MS Excel (95.6 KB)Disclosure of interest
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Li ZY, Wang D, Bian JH, et al. Research progress on the molecular biology of Lotus corniculatus. Pratacult Sci. 2019;36(11):2871–2886.
- Pellissier V, Muratet A, Verfaillie F, et al. Pollination success of Lotus corniculatus (L.) in an urban context. Acta Oecol. 2012;39:94–100.
- Long ZF, Tang CB, Meng JJ, et al. selection of pastures for water and soil conservation in mountainous karst area of Guizhou Province. Guizhou Sci. 2007;25(S1):483–493.
- Sun YX, Wang D, Bai YL, et al. Overexpression of soybean GmNHX1 in lotus root: the decrease of Na + content in body is the basis of improving salt tolerance. Chinese Sci Bull. 2006;51(11):1306–1936.
- Marcus PI, Heide LVD, Sekellick MJ. Interferon action on avian viruses. I. Oral administration of chicken interferon-alpha ameliorates Newcastle disease. J Interferon Cytokine Res. 1999;19(8):881–885.
- Gabriele L, Ozato K. The role of the interferon regulatory factor (IRF) family in dendritic cell development and function. Cytokine Growth Factor Rev. 2007;18(5-6):503–510.
- Wei L, Sandbulte MR, Thomas PG, et al. NFkappaB negatively regulates interferon-induced gene expression and anti-influenza activity. J Biol Chem. 2006;281(17):11678–11684.
- Orchansky P, Rubinstein M, Sela I. Human interferons protect plants from virus infection. Proc Natl Acad Sci USA. 1982;79(7):2278–2280.
- Devash Y, Biggs S, Sela I. Multiplication of tobacco mosaic virus in tobacco leaf disks is inhibited by (2'-5) oligoadenylate. Science. 1982;216(4553):1415–1416.
- Van L. Induced resistance in plants and the role of pathogenesis-related proteins. Eur J Plant Pathol. 1997;103(9):753–765.
- El-Ayouty Y, El-Manawy I, Nasih S, et al. Engineering Chlamydomonas reinhardtii for expression of functionally active human interferon-α. Mol Biotechnol. 2019;61(2):134–144.
- Razmi S, Javaran MJ, Bagheri A, Biotechnology and Plant Breeding Department, Agriculture Faculty, Tarbiat Modares University, Tehran, Iran, et al. Expression of human interferon gamma in tobacco chloroplasts. Rom Biotechnol Lett. 2019;24(2):208–215.
- Zhu Z, Li X, Sun Y, et al. Plant regeneration of transgenic rice and expression of exogenous human α-interferon cDNA. Sci China Ser B. 1992;2:149–155.
- Sawahel W. The production of transgenic potato plants expressing human a-interferon using lipofectin-mediated transformation. Cell Mol Biol Lett. 2002;7(1):19–29.
- Ohya K, Matsumura T, Itchoda N, et al. Ability of orally administered IFN-alpha-containing transgenic potato extracts to inhibit Listeria monocytogenes infection. J Interferon Cytokine Res. 2005;25(8):459–466.
- Zhu Z, Hughes KW, Huang L, et al. Expression of human α-interferon cDNA in transgenic rice plants. Plant Cell Tiss Organ Cult. 1994;36(2):197–204.
- Jiang MC, Hu CC, Lin NS, et al. Production of human IFNγ Protein in Nicotiana benthamiana plant through an enhanced expression system based on bamboo mosaic virus. Viruses. 2019;11(6):509.
- Sekellick MJ, Ferrandino AF, Hopkins DA, et al. Chicken interferon gene: cloning, expression, and analysis. J Interferon Res. 1994;14(2):71–79.
- Sela I. Interferon-like factor from virus-infected plants. Presp Virol. 1981;11:129–139.
- Song L, Zhao D, Tian X, et al. Expression of ChIFN-α in transgenic tobacco causes resistance to tobacco mosaic virus. J Agric Sci Tech Iran. 2010;12(1):118–122.
- Pandi-Perumal SR, Trakht I, Srinivasan V, et al. Physiological effects of melatonin: role of melatonin receptors and signal transduction pathways. Prog Neurobiol. 2008;85(3):335–353.
- Chen H, Feng Y, Zhou J, et al. Research advance of plant root biology. World For Res. 2013;26(5):25–29.
- Lakehal A, Bellini C. Control of adventitious root formation: insights into synergistic and antagonistic hormonal interactions. Physiol Plant. 2019;165(1):90–100.
- Bollmark M, Kubát B, Eliasson L. Variation in endogenous cytokinin content during adventitious root formation in pea cuttings. J Plant Physiol. 1988;132(3):262–265.
- Li S-W, Shi R-F, Leng Y. De novo characterization of the mung bean transcriptome and transcriptomic analysis of adventitious rooting in seedlings using RNA-Seq. PLoS One. 2015;10(7):e0132969.
- Villacorta-Martín C, Sánchez-García AB, Villanova J, et al. Gene expression profiling during adventitious root formation in carnation stem cuttings. BMC Genomics. 2015;16(1):789
- Libao C, Minrong Z, Zhubing H, et al. Comparative transcriptome analysis revealed the cooperative regulation of sucrose and IAA on adventitious root formation in lotus (Nelumbo nucifera Gaertn). BMC Genomics. 2020;21(1):653.
- Tian Z, Hong H, Kang X, et al. Transcriptome analysis of the effect of cutting age on adventitious root formation of populus. Bot Res. 2019;08(03):293–306.
- Wang J, Xia J, Xiao HL, et al. Study on the ecological and mechanical properties of root system of slope protecting plants. J Hubei Univ Technol. 2019;34(4):77–80.
- Subramaniyam S, Mathiyalagan R, Natarajan S, et al. Transcript expression profiling for adventitious roots of Panax ginseng Meyer. Gene. 2014;546(1):89–96.
- Chen K, Guo B, Yu C, et al. Comparative transcriptome analysis provides new insights into the molecular regulatory mechanism of adventitious root formation in Ramie (Boehmeria nivea L.). 2020. DOI. 10.21203/rs.3.rs-23123/v1.
- Du W, Ban Y, Nie H, et al. A comparative transcriptome analysis leads to new insights into the molecular events governing root formation in mulberry softwood cuttings. Plant Mol Biol Rep. 2016;34(2):365–373.
- Fukuda Y, Hirao T, Mishima K, et al. Transcriptome dynamics of rooting zone and aboveground parts of cuttings during adventitious root formation in Cryptomeria japonica D. Don. BMC Plant Biol. 2018;18(1):201–214.
- Song L, Zhao DG. Optimization of culture conditions for Lotus japonicus regeneration in vitro. Mol Plant Breed. 2015;13(04):910–914.
- Song L, Zhao DG, Wu YJ. Transient expression of chicken alpha interferon gene in lettuce. J Zhe Jiang Univ Sci B. 2008;9(5):351–355.
- Wang Y, Song F, Zhu J, et al. GSA: Genome Sequence Archive. Genom Proteom Bioinf. 2017;15(1):14–18.
- Zhang Z, Zhao W, Xiao J, et al. Database Resources of the National Genomics Data Center in 2020. Nucleic Acids Res. 2020;48(D1):D24-D33.
- Hartley SW, Mullikin JC. QoRTs: a comprehensive toolset for quality control and data processing of RNA-Seq experiments. BMC Bioinform. 2015;16(1):1–7.
- Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21.
- Anders S, Pyl PT, Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
- Yu G, Wang LG, Han Y, et al. clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS: A Journal of Integrative Biology. 2012;16(5):284–287.
- Zhang T, Song C, Song L, et al. RNA sequencing and coexpression analysis reveal key genes involved in α-linolenic acid biosynthesis in Perilla frutescens seed. IJMS. 2017;18(11):2433.
- Zheng Y, Jiao C, Sun H, et al. iTAK: a program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol Plant. 2016;9(12):1667–1670.
- Smoot ME, Ono K, Ruscheinski J, et al. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011; 27(3):431–432.
- Untergasser A, Cutcutache I, Koressaar T, et al. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012;40(15):e115.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta c(T)) method. Methods. 2001;25(4):402–408.
- Chen C, Chen H, Zhang Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13(8):1194–1202.
- Guang Q, Song L, Zhao DG. Effect of low temperature stress on physiological responses of transgenic ChIFN-α lotus plant (Lotus corniculatus L.) to cold. Mol Plant Breed. 2015;13(12):2849–2853.
- Wu YJ, Zhao DG, Zhan SN, et al. A study on the insect-resistant mechanism of ChIFN-γ transgenic tobacco. J Yunnan Univ. 2010;32(4):473–479.
- Pop TI, Pamfil D, Bellini C. Auxin control in the formation of adventitious roots. Not Bot Hort Agrobot Cluj. 2011;39(1):307–316.
- Chanclud E, Lacombe B. Plant hormones: key players in gut microbiota and human diseases? Trends Plant Sci. 2017;22(9):754–758.
- Shang C, Yang H, Ma S, et al. Physiological and transcriptomic changes during the early phases of adventitious root formation in mulberry stem hardwood cuttings. IJMS. 2019;20(15):3707.
- Ellenberg J, Swedlow JR, Barlow M, et al. A call for public archives for biological image data. Nat Methods. 2018;15(11):849–854.
- Varaud E, Brioudes F, Szécsi J, et al. Auxin response factor8 regulates Arabidopsis petal growth by interacting with the bHLH transcription factor BIGPETALp. Plant Cell. 2011;23(3):973–983.
- Yin X, Yi K, Zhao Y, et al. Revealing the full-length transcriptome of Caucasian clover rhizome development. BMC Plant Biol. 2020;20(1):429.
- Mu J, Tan H, Hong S, et al. Arabidopsis transcription factor genes NF-YA1, 5, 6, and 9 play redundant roles in male gametogenesis, embryogenesis, and seed development. Mol Plant. 2013;6(1):188–201.
- Zanetti ME, Rípodas C, Niebel A. Plant NF-Y transcription factors: key players in plant–microbe interactions, root development and adaptation to stress. Biochim Biophys Acta Gene Regul Mech. 2017;1860(5):645–654.
- Swain S, Myers ZA, Siriwardana CL, et al. The multifaceted roles of NUCLEAR FACTOR-Y in Arabidopsis thaliana development and stress responses. Biochim Biophys Acta Gene Regul Mech. 2017;1860(5):636–644.
- Li A, Lakshmanan P, He W, et al. Transcriptome profiling provides molecular insights into auxin-induced adventitious root formation in sugarcane (Saccharum spp. interspecific hybrids) Microshoots. Plants. 2020;9(8):931.
- Lee SI, Muthusamy M, Nawaz MA, et al. Genome-wide analysis of spatiotemporal gene expression patterns during floral organ development in Brassica rapa. Mol Genet Genomics. 2019;294(6):1403–1420.
- Tong J, Chen J, Zhu J, et al. Effects of rice C2C2 zinc finger protein gene OsLSD2 on the growth and nitrogen utilization of nipponbare. Chinese J Rice Sc. 2014;28(4):435–441.
- Chen K, Li H, Chen Y, et al. TaSCL14, a novel wheat (Triticum aestivum L.) GRAS gene, regulates plant growth, photosynthesis, tolerance to photooxidative stress, and senescence. J Genet Genomics. 2015;42(1):21–32.
- Kong X, Tian H, Yu Q, et al. PHB3 maintains root stem cell niche identity through ROS-responsive AP2/ERF transcription factors in Arabidopsis. Cell Rep. 2018;22(5):1350–1363.
- Upadhyay S, Jeena GS, Kumar S, et al. Asparagus racemosus bZIP transcription factor-regulated squalene epoxidase (ArSQE) promotes germination and abiotic stress tolerance in transgenic tobacco. Plant Sci. 2020;290:110291.
- Wang L, Xu Y, Zhang C, et al. OsLIC, a novel CCCH-type zinc finger protein with transcription activation, mediates rice architecture via brassinosteroids signaling. PloS One. 2008;3(10):e3521
- Chai G, Kong Y, Zhu M, et al. Arabidopsis C3H14 and C3H15 have overlapping roles in the regulation of secondary wall thickening and anther development. J Exp Bot. 2015;66(9):2595–2609.
- Sun Q, Huang J, Guo Y, et al. A cotton NAC domain transcription factor, GhFSN5, negatively regulates secondary cell wall biosynthesis and anther development in transgenic Arabidopsis. Plant Physiol Biochem. 2020;146:303–314.
- Li T, Yang S, Kang X, et al. The bHLH transcription factor gene AtUPB1 regulates growth by mediating cell cycle progression in Arabidopsis. Biochem Biophys Res Commun. 2019;518(3):565–572.
- Safi A, Medici A, Szponarski W, et al. The world according to GARP transcription factors. Curr Opin Plant Biol. 2017;39:159–167.
- Shen X, Hu Z, Xiang X, et al. Overexpression of a stamen-specific R2R3-MYB gene BcMF28 causes aberrant stamen development in transgenic Arabidopsis. Biochem Biophys Res Commun. 2019;518(4):726–731.
- Yu LH, Miao ZQ, Qi GF, et al. MADS-box transcription factor AGL21 regulates lateral root development and responds to multiple external and physiological signals. Mol Plant. 2014;7(11):1653–1669.
- De Folter S, Shchennikova AV, Franken J, et al. A Bsister MADS-box gene involved in ovule and seed development in petunia and Arabidopsis. Plant J. 2006;47(6):934–946.
- Ripoll JJ, Roeder AH, Ditta GS, et al. A novel role for the floral homeotic gene APETALA2 during Arabidopsis fruit development. Development. 2011;138(23):5167–5176.
- Laity JH, Lee BM, Wright PE. Zinc finger proteins: new insights into structural and functional diversity. Curr Opin Struct Biol. 2001;11(1):39–46.
- Segal DJ, Stege JT, Barbas IIC. Zinc fingers and a green thumb: manipulating gene expression in plants. Curr Opin Struct Biol. 2003;6(2):163–168.