
Abstract
Garcinia paucinervis is an evergreen tree with high medicinal value. Due to its vulnerable reproductive capacity coupled with excessive logging by humans, G. paucinervis has become an endangered species. In order to protect this species effectively, we drew the complete chloroplast genome of G. paucinervis and performed a series of comparative analyses on G. paucinervis and its neighbouring species. The chloroplast genome size of G. paucinervis is 157702 bp. In the chloroplast genome, we identified 130 genes, 56 RNA editing sites in the protein-coding genes, as well as 241 simple sequence repeats (SSRs) and 49 complex repetitive sequences. Comparative analysis identified some high divergent sequences in the intergenic spacers, which can be used as candidate markers for phylogenetic study. From an adaptive evolution point of view, a branch-site model analysis identified positively selected sites in 5 genes, most of which are involved in ribosome biogenesis, protein synthesis and other developmental processes. The detected codon substitutions may be associated with the evolution of G. paucinervis to adapt to the extreme habitat in karst. In addition, the result of the phylogenetic analysis supports the previous phylogenomic studies of taxa within the rosids clade. The ML tree revealed that G. mangostana was a sister to G. gummi-gutta, and they formed a diverging lineage to G. paucinervis in Garcinia. The above information is of great significance for us to understand the evolution of G. paucinervis chloroplast and lays the foundations for future studies in species conservation.
Introduction
Garcinia paucinervis (Garcinaceae) is a precious timber tree species which grows only in the karst mountain in southwestern China and northern Vietnam. This tree exhibits strong and heavy wood material characteristics, which make it suitable for house construction, furniture manufacture and ship-building [Citation1]. In addition to the economic value, the branches, leaves and bark of G. paucinervis were traditionally used as medicine for heat-clearing, detoxification and detumescence [Citation2]. At present, due to its vulnerable reproductive capacity coupled with excessive logging by humans, G. paucinervis has become an endangered species and has been listed as a class II national key protected species in China [Citation3]. In order to protect this species effectively, researchers have paid a lot attention to formulate strategies for the cultivation and conservation of G. paucinervis [Citation4]. However, there are still few studies regarding the evolution and phylogeny of G. paucinervis on the molecular level, which we believe to be an important yet underappreciated direction for the conservation of this species.
The chloroplast is a plant-specific organelle and the main place for photosynthesis. In angiosperms, the chloroplast genome is one of the three DNA genomes, which belongs to maternal inheritance and has highly conservative characteristics. The chloroplast genome of traditional angiosperms ranges from 120 and 160 kb in length [Citation5, Citation6], and is a circular structure composed of four regions (i.e. a pair of inverted repeats (IRs), a large single-copy region (LSC) and a small single-copy region (SSC)). The contraction and expansion of the IR region is the main reason for the mutation of the chloroplast genome, but in general, the chloroplast genome is conserved in gene content and organization, this characteristic make it an ideal genetic resource for plant evolutionary studies [Citation7]. Recent studies demonstrated that the chloroplast genome has the potential to resolve phylogenetic relationships at the species level, infer divergence dating and elucidate the adaptive evolution in specific species [Citation8]. Especially, the molecular markers derived from the chloroplast genome can be used to determine the gene flow, genetic structure and genetic diversity among different populations [Citation9]. Such valuable information can facilitate the efficient formulation of strategies for the conservation of plant genetic resources, and ultimately minimize the extinction risk of endangered plants.
In this study, we assembled the complete chloroplast genome of G. paucinervis and characterized its genomic structure and features including the gene content, repeat sequence and IR expansions/contractions, etc. Combining the previously reported chloroplast genomes of related species, we analyzed the sequence divergence, phylogenetic relationship and adaptive evolution. These results will provide insights into the RNA editing patterns, positively selected sites and evolutionary history of G. paucinervis, and the highly divergent regions detected could be employed to develop potential markers for plant identification. This information will also help us to develop an effective strategy for the conservation of G. paucinervis.
Materials and methods
Plant materials and DNA extraction
Fresh leaves of G. paucinervis were collected from Guangxi Institute of Botany, Chinese Academy of Sciences, Guilin, China. The total genomic DNA was extracted according to the modified CTAB method [Citation10]. Since the G. paucinervis leaf contains high level of polysaccharides and polyphenols, we used double dosage of polyvinylpyrrolidone and β-mercaptoethanol during the early stages of DNA extraction. Finally, the DNA was quantified using a Nanodrop 2000 spectrophotometer.
Genome sequencing and assembling
The DNA of sufficient quality was fragmented by mechanical breakage, and then a series of treatments such as fragment purification and terminal repair were carried out. The fragments of 350 bp in length were selected via agarose gel electrophoresis and then amplified by polymerase chain reaction (PCR) to construct a sequencing library [Citation11]. A paired-end library was constructed using Illumina NovaSeq platform following the manufacturer’s protocol (Illumina, San Diego, California, USA). After quality assessment, the chloroplast genome related reads were filtered by mapping all the raw reads to the reference chloroplast genome of Garcinia mangostana (NC_036341). The filtered reads were assembled into contigs using SPAdes software (version: 3.10.1; parameter:-k 127) [Citation12]. All the contigs were aligned to the reference chloroplast genome by blastn (version: BLAST 2.2.30+; parameter: -evalue 1e-5) [Citation13]. Based on the alignment result, the gap between contigs was filled by Gapcloser included in the SOAP package v1.12 [Citation14]. Finally, the complete chloroplast genome sequence of G. paucinervis was obtained.
Genome annotation and sequence characterization
The chloroplast genome was annotated using the online program GeSeq, a special annotation software for organelles [Citation15]. The coding sequences (CDS) and rRNAs were identified based on BLAST homology searches, followed by manual correction for the questionable annotation. Meanwhile, all tRNA genes were confirmed using the tRNAscan-SE online search server. The circular genome map was drawn by the OGDRAW program [Citation15] and the whole sequence was submitted to GenBank (MT501656). The GC content was calculated for the whole chloroplast genome, and the codon usage frequencies and the relative synonymous codon usage (RSCU) were analyzed for all the CDS using MEGA5.1. In addition, potential RNA editing sites in protein-coding genes were predicted using the PREP suite with a cut-off value of 0.8 [Citation16]. To identify simple sequence repeats (SSRs), the Perl script MISA was used with the filter thresholds set at repeat units ≥ 8 for mononucleotide, repeat units ≥ 4 for dinucleotides and trinucleotides, and repeat units ≥ 3 for tetranucleotides, pentanucleotides and hexanucleotides. All of the SSRs found were manually checked, and the redundant results were discarded. For the identification of complex repeative sequences (i.e. forward, reverse, complement and palindromic), REPuter online software was used with parameters set as follows: hamming distance equal to 3, the minimum repeat size ≥ 30 bp and the sequence consistency of ≥ 90% [Citation17].
Sequence divergence and positive selected analysis
To explore the divergence hotspot regions that facilitate the identification of closely related species, the mVISTA online software was used to compare the chloroplast genome of G. paucinervi with those of G. mangostana, G. gummi-gutta, C. cochinchinense and M. ferrea. This comparative analysis was carried out in shuffle-LAGAN mode with the annotation of G. paucinervi as reference, and the sequence alignment was visualized in an mVISTA plot [Citation18]. To better visualize the expansion/contraction event in IR region, IRscope online software was used to compare the junction regions of IR-LSC and IR-SSC in the chloroplast genome of these five species [Citation19]. G. paucinervi is a special plant endemic to karst environment. To detect whether plastid genes in G. paucinervi were under positive selection, non-synonymous (dN) and synonymous (dS) substitution rates as well as the dN/dS (ω) values of protein-coding genes were calculated in this study. All of the CDS sequences were extracted from G. paucinervi and another 15 species in Malpighiales, and then the single-copy CDS sequences shared by all the species were selected and aligned using the codon model. After alignments, a branch-site model and Bayesian Empirical Bayes (BEB) method were employed to test the potential positive selection of a single codon in G. paucinervi chloroplast genome using PAML v4.8 [Citation20]. Selective pressure was measured by the ratio (ω): ω > 1 indicates positive selection, ω = 1 indicates neutral selection, whereas ω < 1 indicates negative selection. In the test of the branch-site model, the branch under test for positive selection was set as the foreground branch (G. paucinervi, G. mangostana and G. gummi-gutta), while the other branches were set as background branches (all the other tested species). Log likelihood values of branch-site models as well as the statistical significance were calculated using likelihood ratio tests (LRT). Furthermore, BEB method was used to test the posterior probability of amino acid sites to identify the positively selected sites. Finally, the amino acid sequences of positively selected genes were displayed using the PSIPRED server (http://bioinf.cs.ucl.ac.uk/psipred).
Phylogenetic analysis
For the phylogenetic analysis, 17 species representing different orders in the rosids lineage were recruited to construct the phylogenetic tree. Seventy-eight protein-coding genes were extracted from the chloroplast genome of the analyzed species, and then aligned using MAFFT (version 7.222) with the default parameters [Citation21]. We concatenated these gene sequences into a supermatrix and constructed the phylogenetic tree using Maximum Likelihood (ML) method by RAxML 7.0.4 [Citation22] with 1000 replicates under the GTR + CAT model. Vitis vinifera was designated as the out-group.
Results
Chloroplast genome organization of G. paucinervis
Like traditional angiosperms, the chloroplast genome of G. paucinervis consists of four regions: LSC, SSC and a pair of IRs (). The total length of the G. paucinervis chloroplast genome is 157702 bp. The length of the LSC region is 85989 bp, which is the longest among the four regions; the second longest is the IR region, which is 26988 bp; and the smallest is the SSC region with a length of 17737 bp (). In the whole chloroplast genome, adenine (A), thymine (T), guanine (G) and cytosine (C) account for 31.61%, 32.18%, 17.76% and 18.45%, respectively. In addition, the GC content in the IR region is 42.30%, and it is 33.70% in the LSC region, while the GC content in the SSC region is 30.40%. We also noted that the GC content of rRNA and tRNA genes is 55.6% and 53.1%, whereas the GC content of the CDS is just 36.8%. Because all the rRNA genes are only distributed in the IR regions, this is one of the reasons that contribute to the highest GC content in the IR region.
Figure 1. Circular map of the chloroplast genome of G. paucinervis. Genes belonging to different functional groups are highlighted in different colours. Genes on the outside of the circle are transcribed counter-clockwise, while genes on the inside are transcribed clockwise. The darker grey in the inner circle corresponds to GC content, and the lighter grey corresponds to AT content.
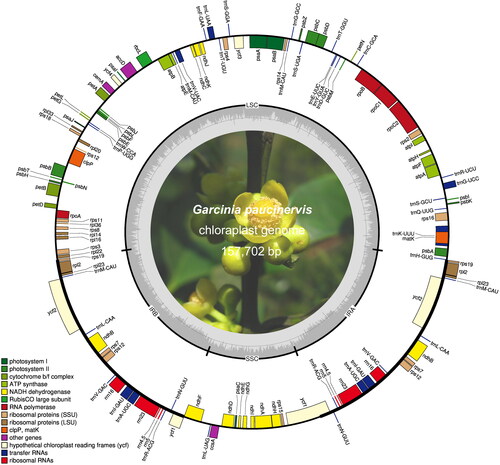
Table 1. Summary of the chloroplast genomes of G. paucinervis and its neighbouring species.
A total of 130 genes were identified in the chloroplast genome of G. paucinervis, including 85 protein-coding genes, 37 tRNA genes and 8 rRNA genes. Among them, there are 94 unique genes, whereas a total of 18 genes are duplicated in the chloroplast genome, including 6 CDS genes, 8 tRNA genes and 4 rRNA genes. Each gene in the chloroplast genome has a specific function [Citation23]. There are 44 genes involved in photosynthesis, which include six genes for ATP synthase, eleven genes for NADH dehydrogenase, six genes for the cytochrome b/f complex, five genes for photosystem I, fifteen genes for photosystem II, and one gene for the large chain of Rubisco (). In addition, there are 29 genes encoding tRNA, 12 genes encoding small ribosomal proteins (SSU), 8 genes encoding large ribosomal proteins (LSU), 4 genes encoding rRNA and 4 genes encoding RNA polymerase in chloroplast genome. We also identified four hypothetical chloroplast reading frames (ycf), two genes related to enzyme synthesis (matK and clpP) and three other genes (cemA, accD and ccsA) in the chloroplast genome.
Table 2. Genes in the chloroplast genome of G. paucinervis.
There are 18 genes with introns in the chloroplast genome of G. paucinervis (). Among these genes, there are 5 tRNA genes and 13 protein-coding genes. We found that rps12 locates its first exon in the LSC region and two other exons in the IRs. This phenomenon was also found in other angiosperms [Citation24]. The comparative analysis results showed that the trnK-UUU gene has the longest intron, which is 2552 bp in length; rpoC1 has the longest exon and its length is 1634 bp. According to statistics, except the clpP gene, which has two introns, the other 17 genes have only one intron.
Table 3. Summary of genes with introns in G. paucinervis chloroplast genome.
We compared the chloroplast genome of G. paucinervis with that of four neighbouring species (G. mangostana, G. gummi-gutta, C. cochinchinense, M. ferrea) and found that among the five complete chloroplast genomes, the longest is M. ferrea, which is 161470 bp in length, and it is longer than that of G. paucinervis by 3850 bp. The size of the chloroplast genome among these plants is M. ferrea > G. mangostana > G. paucinervis > C. cochinchinense > G. gummi-gutta. We noted that the total GC content of the five species is similar, but M. ferrea still has the largest GC content, which is 36.42%. Then we analyzed the gene number of chloroplast genome of five species, and the results showed that there is little difference in the number of CDS, the number of tRNA genes and the number of rRNA genes ().
Codon usage and RNA editing in protein coding genes
Codon is a sequence of three nucleotides which together form a unit of genetic code in DNA or RNA molecule. Although one codon can only correspond to a specific amino acid, a specific amino acid can correspond to more than one codon [Citation25]. In this study, we found that there are 23337 codons (including stop codons) in the chloroplast genome of G. paucinervis and 20 amino acids are translated. According to statistics, leucine is the most abundant amino acid and it takes up 2456 codons, followed by isoleucine with 2054 codons. The least prevalent amino acid is cysteine, which only takes up 268 codons. Subsequently, we used MEGA5 software to analyze the relative synonymous codon usage (RSCU) in the G. paucinervis chloroplast genome. In principle, when the RSCU value of a codon is > 1, it is considered to be a relatively frequently used codon. In contrast, an RSCU value < 1 means that the codon is a relatively less used codon. The RSCU value equal to 1 indicates there is no preference for codon use [Citation26]. We found that there are 64 types in these 23337 codons. Among them, 30 types of codons have RSCU values > 1, and these codons all end with A or U except UUG (). There are 32 types of codons whose RSCU values are < 1, and most of these codons end with G or C. In addition, the RSCU values of AUG and UGG are equal to 1, which suggests that the two types of codons have no bias in expression.
Table 4. The codon–anticodon recognition pattern and codon usage in the chloroplast genome of G. paucinervis.
Generally, the initiation codon of the protein coding gene is ATG, but we found some differences when checking all the CDS. We found that the initiation codon of the ndhD gene is ACG rather than ATG. At first we thought it is a special case of RNA editing, which can convert ACG into a standard ATG initiation codon. As expected, some previous studies have conducted related experiments on whether ACG could serve as the initiation codon, and their results demonstrated that the chloroplast translator accepts ACG use as an initiation codon [Citation27]. Although the protein-coding genes seldom use ACG as the start codon, there are still many genes that use ACG as the initiation codon for translation [Citation28]. Therefore, it is not a sequencing error in this case that the ndhD gene uses ACG as the initiation codon.
To examine whether there are any RNA editing sites in other genes, we used the PREP suite to perform RNA editing prediction for all the protein-coding genes in the G. paucinervis chloroplast genome by setting the first codon position of the first nucleotide. Consequently, a total of 56 RNA editing sites are identified in the G. paucinervis chloroplast genome (), and these sites were changed from C-to-U. The ndhB gene has 11 RNA editing sites, and this is the gene that contains the largest number of RNA editing sites among all the protein-coding genes. The second is the ndhD gene, which contains 10 RNA editing sites. Further analysis found that most of the RNA editing events occurred to change polar amino acids to non-polar amino acids. However, each of the genes matK, petD, ndhD and psbE, has an RNA editing site where conversion changed from non-polar amino acids to polar amino acids.
Repeat sequence analysis
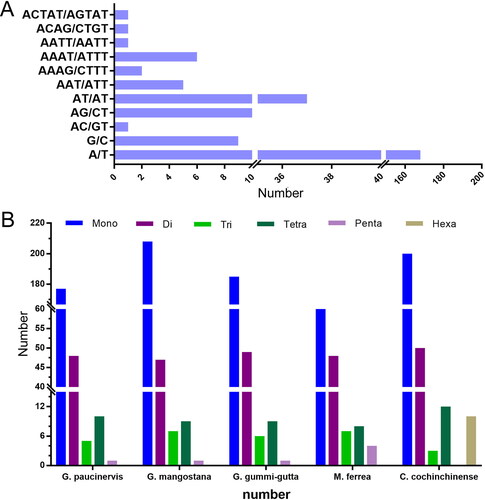
SSRs are tandemly repeated DNA sequences widely distributed in eukaryote genomes [Citation29]. Nowadays, SSRs have become one of the robust molecular markers for plant genetic analysis. Through statistical analysis, a total of 241 SSRs were found in the chloroplast genome of G. paucinervis (). Among the 241 simple sequence repeats, there are 177 mononucleotide repeats, 48 dinucleotide repeats, 5 trinucleotide repeats, 10 tetranucleotide repeats and one pentanucleotide, but no hexanucleotides were found. Among all the mononucleotide repeats, the amount of A/T is much higher than that of G/C, and it is also the highest among all SSRs, accounting for 69.71% of all the SSRs. This result is consistent with the fact that the complete chloroplast genome of G. paucinervis is rich in A/T. In all SSRs, the most frequent repeats are A/T, followed by AT/AT, AG/CT, G/C, AAAT/ATTT, AAT/ATT and AAAG/CTTTT. We also found that there are four SSRs that appear only once. They are AC/GT, AATT/AATT, ACAG/CTGT and ACTAT/AGTAT. Subsequently, we conducted a comparative analysis of SSRs in G. paucinervis, G. mangostana, G. gummi-gutta, C. cochinchinense and M. ferrea. The results showed that C. cochinchinense has the largest number of SSR, which is 275. M. ferrea has the least number of SSR among the five species (). In addition, G. mangostana has the largest number of mononucleotide repeats, while C. cochinchinense has the largest number of dinucleotide repeats. Both G. mangostana and M. ferrea have seven trinucleotide repeats, and they also have the most trinucleotide repeats among the five species. C. cochinchinense and M. ferrea have the largest number of tetranucleotide and pentanucleotides repeats, respectively. Notably, ten hexanucleotide repeats were found in the C. cochinchinense chloroplast genome, but not in the other four species. The identified SSRs could be used to evaluate the genetic diversity and population structure pattern of these species.
Figure 2. Analysis of simple sequence repeats (SSRs) in five chloroplast genomes. (A) The type and quantity of SSRs in the chloroplast genome of Garcinia paucinervis. (B) Comparative analysis of SSRs in five chloroplast genomes.
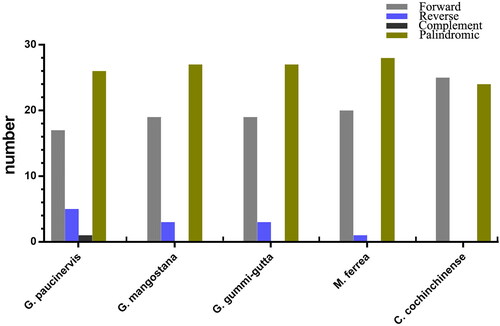
In addition to SSRs, we also explored the long complex repetitive sequences including forward repeats, reverse repeats, palindromic repeats and complement repeats in the G. paucinervis chloroplast genome. Repetitive regions are considered to play an important role in genome recombination and rearrangement, which could lead to the variation of species. A total of 49 repetitive structures were found in the G. paucinervis chloroplast genome, including 17 forward repeats, 26 palindromes, five reverse repeats and one complement (). Among them, there are 29 repeative sequences (15 palindromic repeats, 8 forward repeats, 5 reverse repeats and one complement repeat) in the LSC region, 5 repetitive sequences (three palindromic repeats and two forward repeats) in the SSC region, and 15 repetitive sequences (8 palindromic repeats and 7 forward repeats) in the two IR regions. We also analyzed the repetitive sequences of four neighbouring species (G. mangostana, G. gummi-gutta, C. cochinchinense and M. ferrea), and identified a total of 49, 49, 49, 49 repeat structures in these species, including 19, 19, 25, 20 forward repeats, 27, 27, 24, 28 palindromic repeats, 3, 3, 0, 1, reverse repeats and 0, 0, 0, 0 complement repeats, respectively.
Figure 3. Identification and comparison of complex repetitive sequences in five chloroplast genomes.
Sequence divergence among different species
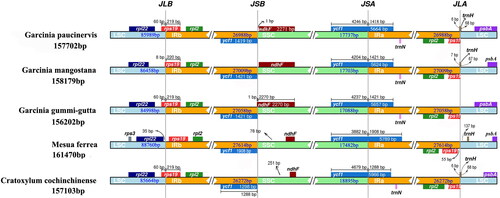
The IR expansion and contraction have contributed to the size difference of chloroplast genome and are considered as a major factor affecting plant evolution. We compared the IR boundaries of five species using IRscope online software. There are obvious differences in the SC/IR boundaries among the five chloroplast genomes (). In combination with the annotation files of chloroplast genomes, we found that there are four genes located at or adjacent to the boundaries of LSC/IR and SSC/IR, including ndhF, ycf1, rps19 and trnH, and these genes vary in size and location among different species. Although all of the chloroplast genomes of these species have two rps19 genes, only M. ferrea conserved its rps19 gene entirely in IR regions, and separated from the IR/LSC region by 55 bp. The IR region of other four species contracted into rps19 gene, resulting in a pseudogene fragment with lengths of 219-220 bp at the IRa/LSC border. In addition, although the rps19 gene of G. mangostana also crossed the IRb/LSC junction, some deletion of gene fragments has occurred in this gene obviously, resulting in approximately 50 bp shorter length than that in other species. Similarly to rps19, the SSC/IRa border was located in the ycf1 gene, creating a long ycf1 pseudogene fragment in all of the species. The length of the ycf1 pseudogene was 1419 bp in G. paucinervis, 1421 bp in G. mangostana and G. gummi-gutta, 1908 bp in M. ferrea and 1298 bp in C. cochinchinense. Besides, the ycf1 pseudogene and the ndhF gene overlapped in G. gummi-gutta chloroplast genome by 1 bp, while in the other four species, the ndhF gene was located entirely at the SSC region. We also noted that the trnH gene crossed the LSC/IRa border and overlapped with the rps19 pseudogene in G. paucinervis, G. mangostana and C. cochinchinense, while it was fully located in the IRa region in M. ferrea 137 bp away from the IRa/LSC border. As indicated above, we can conclude that the variations of SC/IR boundaries have led to the length variation of the whole chloroplast genome.
Figure 4. Comparison of border positions of LSC, SSC and IRs among five species.
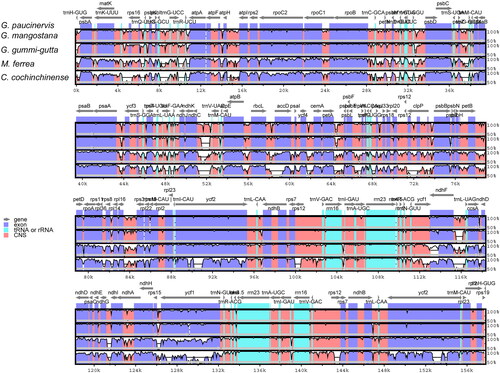
Highly variable regions in coding and non-coding regions together constitute potential DNA barcodes or molecular markers, which could provide basis for phylogenetic analysis and species identification [Citation24]. To identify the high divergence sequences, we used mVISTA online software to analyze the variation in chloroplast genome sequences of five species. The alignment showed that the five chloroplast genome sequences were relatively conservative, but there are some regions that exhibited high sequence divergence (). The sequences in the LSC and SSC regions were more divergent than the sequences in the IR regions. This result is due to the conservative characteristics of rRNA genes, which are all distributed in the IR region. In addition, the sequences of the non-coding regions are also more divergent than that of the coding region. The most divergent non-coding regions among these chloroplast genomes were trnH-psbA, trnT-psbD, psaA-ycf3, ndhC-trnV and ndhD-ccsA.
Figure 5. Sequence comparison of five chloroplast genomes in mVISTA using G. paucinervis as a reference. Arrows indicate the annotated genes and their transcriptional direction. The vertical scale represents the percent identity.
Survey for positive selection in G. paucinervis chloroplast genome
In the chloroplast genome, there are some divergences in protein-coding genes under the influence of natural environment. To examine whether those divergent sites were under positive selection, we used the branch-site model implemented in PAML [Citation20]. Of the 76 protein-coding genes we tested, signals of positive selection were detected in 5 genes (matK, psbK, rps4, rps12 and rps19) with p < 0.05 and a high posterior probability in the BEB test (), indicating that these genes maybe play a key role in the adaptive evolution of G. paucinervis. In the branch-site model, one codon (223rd) in the matK gene was under positive selection with a BEB posterior probability larger than 0.99. The alignment results of the amino acid sequences revealed that although many amino acids vary among different members of different families, the 223rd codon encoding valine (V) was specific for G. paucinervis but instead encoded phenylalanine (F) in other families. The secondary structure of the matK protein predicted by PSIPRED server showed that this positively selected site was located in the α-helix (). In the rps12 gene, the site under positive selection was the codon encoding the 25th amino acid, which is located in the random coil and is valine (V) for G. paucinervis, while glycine (G) in the species from other families (). In addition, we observed that a positively selected site changed from glutamic acid (E) to threonine (T) for G. paucinervis in rps19 (). Interestingly, rps4 was found to harbour two sites under positive selection, which were the 22nd codon encoding alanine (A) and the 117th codon encoding phenylalanine (F) for G. paucinervis, and these sites were located in the random coil and β-sheet respectively (). In the species from other families, the 22nd codon and the 117th codon encoded threonine (T) and leucine (L), respectively. Furthermore, we also detected one codon site under positive selection in psbK, which encodes a protein that directly participates in the photosystem. The observed site was located in the α-helix, and encoded phenylalanine (F) specific for G. paucinervis (), whereas valine (V) was found in the species from other families. It is worth mentioning that most of the positively selected genes are involved in ribosome biogenesis, protein synthesis and other developmental processes.
Figure 6. Secondary structure of matK protein of G. paucinervis. The red box marks the site under positive selection.
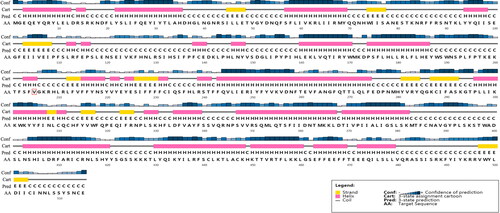
Table 5. Identification of potential positive selection sites based on branch-site model.
Phylogenetic analysis
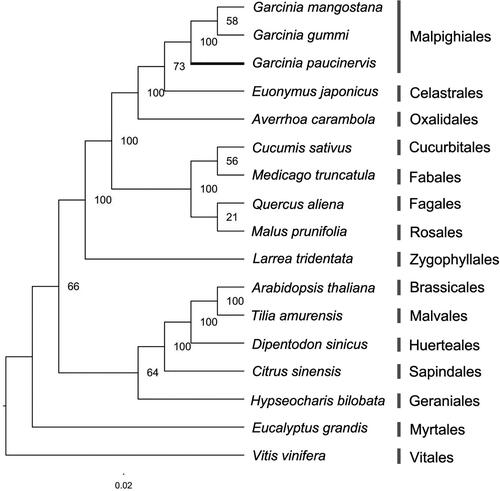
To study the phylogenetic position of G. paucinervis within the rosids lineage, we used 78 protein-coding genes shared by the chloroplast genomes of three species in Garcinia and 14 species from other orders in the rosids lineage to construct an ML tree with V. vinifera as an outgroup. The resulting topology was in accordance with previous phylogeny research [Citation30], suggesting that the phylogenetic tree is reliable. Importantly, the phylogenetic tree revealed that G. mangostana was a sister to G. gummi-gutta, and they contributed to a diverging lineage to G. paucinervis in Garcinia, which is a genus of Garcinaceae in the order Malpighiales (). In addition, the phylogenetic tree continues to support a sister relationship between the Malpighiales and Celastrales, and they were positioned close to Oxalidales. These analyses substantially increase our understanding of the evolutionary relationship among species in the rosids lineage.
Figure 7. ML phylogenetic tree based on 78 protein-coding genes from 17 species of the rosids lineage with V. vinifera as an outgroup. Bootstrap support is shown above branches.
Discussion
The chloroplast genome is known to be highly conserved in gene content and gene order, but the sequence changes in the chloroplast genome, such as gene mutation, repeat sequence rearrangement and the contraction/expansion of IR region border, provide abundant taxonomic data. Up to now, there have been many studies carried out to evaluate the genetic diversity and genetic structure of the endangered plants using DNA markers derived from the chloroplast genome [Citation31–33], demonstrating that the chloroplast genome could provide valuable genetic information for formulating efficient conservation strategies for plant species. In this study, we characterized the complete chloroplast genome of G. paucinervis, an endangered evergreen tree (). Combined with the previously reported chloroplast genomes of four species, we analyzed the differences between these species’ chloroplast genomes. Importantly, we mined 241 SSRs in the chloroplast genome of G. paucinervis, and most of these SSRs were also found in the comparative species but there was a difference in their length. We noted that the majority of di- to hexa- nucleotides are AT-rich in all species. This phenomenon is consistent with the SSR characteristics of chloroplast genomes in angiosperms [Citation34]. All of these SSRs, together with the aforementioned complex repeat sequences, are informative sources for developing markers for genetic diversity studies of G. paucinervis.
RNA editing is a post transcriptional process, which involves the insertion, deletion or conversion of nucleic acid bases in the coding region of the transcribed RNA. The "gene" produced by RNA editing is also called cryptogene, which means that the structure of the product cannot be deduced from the genomic DNA sequence [Citation35, Citation36]. RNA editing is thought to be an important event during the course of plant evolution, since it could introduce the start codon or stop codon in the transcribed RNA to modify the gene expression pattern, and it also increases the amount of coding information of peptide chain; this post-translational modification ultimately confers plants a flexible capacity to respond to the complex environment. Previous studies found that different plant species show a variant degrees of losses in RNA editing sites during evolution [Citation37–39]. In G. paucinervis, a total of 56 RNA editing sites were identified, and all these sites were changes from C-to-U, resulting in the conversion of 12 amino acids (); this result is consistent with previous studies that the C-to-U conversion is the most predominant form in chloroplasts [Citation40, Citation41]. Although U-to-C changes and G-to-A changes were found in other plants [Citation41], we did not detect any of these changes in the G. paucinervis chloroplast. It is possible that these kinds of RNA editing sites were gradually lost in th course of evolution. Further verification on RNA sequencing of chloroplasts will be needed to support this hypothesis. In addition, out of these amino acids, the serine (S) to leucine (L) change was the most common (16/56), followed by serine (S) to phenylalanine (F) change and proline (P) to leucine (L) change (). The ndhB gene has 11 RNA editing sites, which is the gene that contains the largest number of RNA editing sites among the protein-coding genes. This result is also found in Arabidopsis, tobacco, tomato, rice and maize [Citation42, Citation43]. The ndhD is the second most extensively edited gene, which contains 10 RNA editing sites. The ndh genes are known to act as electro-regulators to control the accumulation of reactive oxygen species in cyclic photosynthesis electron transporters [Citation44]. Since G. paucinervis is a drought-tolerant species that grows in karst enviroments, the RNA editing process in ndh genes may help G. paucinervis to more nimbly adjust photosynthesis efficiency when suffering from drought stress. Though the function of RNA editing in the chloroplast has not yet been clearly elucidated, the identified RNA editing sites in our study could help to better understand the physiological behavior of G. paucinervis.
G. paucinervi is a plant endemic to tropical and sub-tropical karst environments. As we know, karst topography is characterized by very poor soil content, low water storage capacity, insufficient light, and nutrient deficiency; these severe conditions could impose a strong selective pressure on plant evolution [Citation45]. To adapt to the extreme habitats, G. paucinervis has evolved with special morphological traits. For example, this plant harbours rigid leaves with thick cuticle to reduce water transpiration; this change may affect the function of some genes in the chloroplast related to photosynthesis and photorespiration [Citation46]. In an attempt to identify whether there exists any positive selection in the chloroplast genes, we identified 5 genes (matK, psbK, rps4, rps12 and rps19) under positive selection using the branch-site model in PAML. One codon (223rd) in the matK gene showed a positive selection signal with a BEB posterior probability larger than 0.99, and this site corresponded to an amino acid located in the α-helix of the encoded protein. The gene matK encodes maturase K, which is involved in group II intron splicing and is essential for RNA processing. Previous studies have proposed that some genes, including atpF, rpl2, rps12, trnA, trnI and trnK, require matK for intron excision in their transcripts, and the protein products from these genes are essential for normal photosynthesis [Citation47], indicating that matK is an indispensable post transcription splicing factor in the chloroplast. Due to its fast substitution rate, matK is also widely used to construct the diverse phylogenetic relationships in higher plants. In addition to matK, we also identified one site under positive selection in psbK, and this site also corresponded to an amino acid located in the α-helix. Interestingly, previous studies did not observe any positive selection signals in this gene in other plants, so this is probably a recent adaptive evolutionary feature in G. paucinervis. Gene psbK encodes a protein that directly participates in photosynthesis. Its protein product is one of the components of the core complex of photosystem II (PSII) which uses light energy to abstract electrons from H2O, generating O2 and a proton gradient subsequently used for ATP formation. It consists of a core antenna complex that captures photons, and an electron transfer chain that converts photonic excitation into a charge separation [Citation48]. The site identified here indicates that the photosystem of G. paucinervis was indeed affected by natural selective forces. rps4, rps12 and rps19 belong to ribosomal protein genes, which encode important proteins for the small ribosome subunit. Previous studies reveal that rps genes encode key players in the decoding center of chloroplast ribosomes and loss of function of a single rps gene could lead to the arrest of cell growth and chloroplast translation [Citation49]. In this respect, the positive selection sites found in the three ribosomal protein genes in the G. paucinervis chloroplast genome could be regarded and further analyzed in a broader perspective. Overall, the positive selection sites found in the five genes reflect a complex adaptive strategy changes in the photosystem likely associated with the adaptation of G. paucinervis to the extreme habitats.
Conclusions
In this study, we characterized the complete choloplast genome of G. paucinervis for the first time. The genome annotation, and comparative analysis showed that the chloroplast genome of G. paucinervis has a typical quadripartite structure like traditional angiosperms, and the GC content, gene number and codon usage features are similar to those of other species in Garcinaceae. The phylogenetic tree supports G. paucinervis belonging to Garcinia in the Clusiaceae family. These data will enlarge our understanding of the evolutionary history of Clusiaceae species. Notably, the SSRs explored here are informative sources for developing markers for genetic diversity studies of G. paucinervis. The adaptive evolution analysis detected signs of positive selection in five chloroplast genes (i.e. matK, psbK, rps4, rps12 and rps19). The change of these adaptive sites may be associated with the evolution of G. paucinervis to adapt to the extreme habitats in karst enviroments. Hence, this information will help us to develop an effective strategy for the conservation of G. paucinervis.
Supplemental Material
Download PDF (677.3 KB)Supplemental Material
Download PDF (183.6 KB)Acknowledgements
We are sincerely grateful to Yusong Huang (Guangxi Institute of Botany, The Chinese Academy of Sciences, Guilin, China) for providing plant picture.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The data generated during this study is available at Genbank (accession number: MT501656)
Additional information
Funding
Related Research Data
References
- Hu G, Zhang ZH, Yang P, et al. Development of microsatellite markers in Garcinia paucinervis (Clusiaceae), an endangered species of karst habitats. Appl Plant Sci. 2017;5(1):1600131.
- Hemshekhar M, Sunitha K, Santhosh MS, et al. An overview on genus Garcinia: phytochemical and therapeutical aspects. Phytochem Rev. 2011; 10(3):325–351.
- Wang S, Xie Y. China Species Red List, 366. Beijing, China: Higher Education Press; 2004.
- Ovalle-Magallanes B, Eugenio-Pérez D, Pedraza-Chaverri J. Medicinal properties of mangosteen (Garcinia mangostana L.): A comprehensive update. Food Chem Toxicol. 2017;109(Pt 1):102–122.
- Nie L, Cui Y, Chen X, et al. Complete chloroplast genome sequence of the medicinal plant Arctiumlappa. Genome. 2020; 63(1):53–60.
- Sabater B. Evolution and function of the chloroplast. Current investigations and perspectives. IJMS. 2018; 19(10):3095.
- Zha X, Wang X, Li J, et al. Complete chloroplast genome of Sophora alopecuroides (Papilionoideae): molecular structures, comparative genome analysis and phylogenetic analysis. J Genet. 2020;99(1):30.
- Cheon KS, Kim KA, Kwak M, et al. The complete chloroplast genome sequences of four Viola species (Violaceae) and comparative analyses with its congeneric species. PLoS One. 2019; 14(3):e0214162
- Zhou T, Ruhsam M, Wang J, et al. The complete chloroplast genome of Euphrasia regelii, pseudogenization of ndh genes and the phylogenetic relationships within. Orobanchaceae Front Genet. 2019; 10:444.
- Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987; 19:11–15.
- Arya M, Shergill IS, Williamson M, et al. Basic principles of real-time quantitative PCR. Expert Rev Mol Diagn. 2005; 5(2):209–219.
- Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012; 19(5):455–477.
- Zhao K, Chu X. G-BLASTN: accelerating nucleotide alignment by graphics processors. Bioinformatics. 2014;30(10):1384–1391.
- Li R, Zhu H, Ruan J, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010; 20(2):265–272.
- Tillich M, Lehwark P, Pellizzer T, et al. GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017; 45(W1):W6–W11.
- Mower JP. JP. The PREP suite: predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009;37(Web Server):W253–W259.
- Kurtz S, Choudhuri JV, Ohlebusch E, et al. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001; 29(22):4633–4642.
- Mayor C, Brudno M, Schwartz JR, et al. VISTA : visualizing global DNA sequence alignments of arbitrary length. Bioinformatics. 2000; 16(11):1046–1047.
- Amiryousefi A, Hyvönen J, Poczai P. IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics. 2018; 34(17):3030–3031.
- Yang Z. Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–1591.
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780.
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690.
- Gui L, Jiang S, Xie D, et al. Analysis of complete chloroplast genomes of Curcuma and the contribution to phylogeny and adaptive evolution. Gene. 2020; 732:144355
- Qian J, Song J, Gao H, et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PloS One. 2013; 8(2):e57607
- Nakamura Y, Wada K, Wada Y, et al. Codon usage tabulated from the international DNA sequence databases. Nucleic Acids Res. 1996; 24(1):214–215.
- Oresic M, Shalloway D. Specific correlations between relative synonymous codon usage and protein secondary structure. J Mol Biol. 1998; 281(1):31–48.
- Zandueta-Criado A, Bock R. Surprising features of plastid ndhD transcripts: addition of non-encoded nucleotides and polysome association of mRNAs with an unedited start codon. Nucleic Acids Res. 2004; 32(2):542–550.
- Hirose T, Sugiura M. Both RNA editing and RNA cleavage are required for translation of tobacco chloroplast ndhD mRNA: a possible regulatory mechanism for the expression of a chloroplast operon consisting of functionally unrelated genes. EMBO J. 1997; 16(22):6804–6811.
- Suwabe K, Tsukazaki H, Iketani H, et al. Simple sequence repeat-based comparative genomics between Brassica rapa and Arabidopsis thaliana: the genetic origin of clubroot resistance. Genetics. 2006; 173(1):309–319.
- Li HT, Yi TS, Gao LM, et al. Origin of angiosperms and the puzzle of the Jurassic gap. Nat Plants. 2019;5(5):461–470.
- Anne HE, Staton ME, Dattilo AJ, et al. Population structure and genetic diversity within the endangered species Pityopsis ruthii (Asteraceae). Front Plant Sci. 2018; 9:943.
- Jian HY, Li SF, Guo JL, et al. High genetic diversity and differentiation of an extremely narrowly distributed and critically endangered decaploid rose (Rosa praelucens): implications for its conservation. Conserv Genet. 2018; 19(4):761–776.
- Terrab A, Talavera S, Arista M, et al. Genetic diversity at chloroplast microsatellites (cpSSRs) and geographic structure in endangered west mediterranean firs (Abies spp. pinaceae). Taxon. 2007; 56(2):409–416.
- Ebert D, Peakall R. Chloroplast simple sequence repeats (cpSSRs): Technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol Ecol Resour. 2009; 9(3):673–690.
- Ichinose M, Sugita M. RNA editing and its molecular mechanism in plant organelles. Genes. 2016; 8(1):5.
- Tulshiram WS. RNA Editing and its Potential Role in Evolution. Biotecharticles2017. Available from: https://www.biotecharticles.com/Microbiology-Article/RNA-Editing-and-its-Potential-Role-in-Evolution-3799.html.
- Adams KL, Qiu YL, Stoutemyer M, et al. Punctuated evolution of mitochondrial gene content: high and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc Natl Acad Sci U S A. 2002; 99(15):9905–9912.
- Maier RM, Zeltz P, Kossel H, et al. RNA editing in plant mitochondria and chloroplasts. Plant Mol Biol. 1996;32(1-2):343–365.
- Mower JP. JP. Modeling sites of RNA editing as a fifth nucleotide state reveals progressive loss of edited sites from angiosperm mitochondria. Mol Biol Evol. 2008;25(1):52–61.
- Bock R. Sense from nonsense: how the genetic information of chloroplasts is altered by RNA editing. Biochimie. 2000; 82(6-7):549–557.
- Tangphatsornruang S, Uthaipaisanwong P, Sangsrakru D, et al. Characterization of the complete chloroplast genome of Hevea brasiliensis reveals genome rearrangement, RNA editing sites and phylogenetic relationships. Gene. 2011;475(2):104–112.
- Kahlau S, Aspinall S, Gray JC, et al. Sequence of the tomato chloroplast DNA and evolutionary comparison of Solanaceous plastid genomes. J Mol Evol. 2006;63(2):194–207.
- Sasaki T, Yukawa Y, Miyamoto T, et al. Identification of RNA editing sites in chloroplast transcripts from the maternal and paternal progenitors of tobacco (Nicotiana tabacum): Comparative analysis shows the involvement of distinct trans-factors for ndhB editing. Mol Biol Evol. 2003; 20(7):1028–1035.
- Ivanova Z, Sablok G, Daskalova E, et al. Chloroplast Genome Analysis of Resurrection Tertiary Relict Haberlea rhodopensis Highlights Genes Important for Desiccation Stress Response. Front Plant Sci. 2017; 8:204.
- Xie DF, Yu Y, Deng YQ, et al. Comparative analysis of the chloroplast genomes of the Chinese endemic genus Urophysa and their contribution to chloroplast phylogeny and adaptive evolution. IJMS. 2018; 19(7):1847.
- Zhao B, Li JJ, Yuan RW, et al. Adaptive evolution of the rbcL gene in the genus Rheum (polygonaceae.). Biotechnol Biotec Eq. 2017; 31(3):493–498.
- Barthet MM, Hilu KW. Expression of matK: functional and evolutionary implications. Am J Bot. 2007; 94(8):1402–1412.
- UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 2015; 43(Database issue):D204-212. Available link: https://www.uniprot.org/uniprot/P60147.
- Ramundo S, Rahire M, Schaad O, et al. Repression of essential chloroplast genes reveals new signaling pathways and regulatory feedback loops in chlamydomonas. Plant Cell. 2013;25(1):167–186.