
Abstract
Flax (Linum usitatissimum L.) is an important industrial crop, and its seeds are rich in a variety of functional nutrition and health components. Therefore, flaxseed is not only an important raw material in agricultural production but also an indispensable part to increase the added value of flax products. To investigate the role of miRNA in the development of flaxseed, the present study used flaxseed of Shuangya 4 at 10, 20 and 30 days post anthesis (DPA) as the study material. The study identified 130 known miRNAs and 93 new miRNAs; a total of 3,361 target genes were predicted, including 2401 target genes corresponding to known miRNAs and 960 target genes corresponding to new miRNAs. Combining the sequencing results of the degradation group, it was found that the known miRNAs including miR156, miR171, miR164, miR408 and miR398 as well as the new miRNA unconservative_scaffold3872_46150 showed significant differential expression in the development of flaxseed. These differentially expressed miRNAs and their target genes were analyzed by quantitative reverse-transcription polymerase chain reaction (qRT-PCR). The results showed that the target genes could be negatively regulated by their corresponding miRNAs and contribute to the development of flaxseed.
Supplemental data for this article is available online at https://doi.org/10.1080/13102818.2021.1903337 .
Introduction
Seed development is one of the most important periods of plant growth, and it not only plays a crucial role in plant colonization in different environments but also provides a major food source for humans [Citation1–3]. Seed development is a very complex biological process involving many metabolic regulatory pathways and biological processes. It can be divided into two main stages: embryo development and seed maturation. The main characteristics are cell division and cell differentiation to form mature embryos in the stage of embryo development. Seed maturation involves the accumulation of various storage materials, for example protein, fat and carbohydrates [Citation4, Citation5]. Analyzing the genetic mechanism of plant seed development can provide an important reference for crop genetic improvement, including regulating seed formation, seed morphology and storage material types. A great deal of work has been done to elucidate the regulatory genes or metabolic pathways related to seed development of various plants in the past few decades, especially model plants of rice [Citation6] and Arabidopsis thaliana [Citation1, Citation7]. These studies identified a large number of key genes for transcriptional regulation or signal transduction. Due to the widespread functional differentiation of direct homologous genes among different plants, the regulatory network of model plant seed development may not be able to fully reveal the metabolism and development of other species.
Most eukaryotes contain RNA-based silencing pathways, whose main function is to mediate gene expression regulation at transcriptional or posttranscriptional levels. Small RNAs are one of the most important members of this silencing system, and they include miRNAs and small interfering RNAs (siRNAs), which participate in gene expression regulation through posttranscriptional regulation or epigenetic modification, respectively [Citation8–10]. miRNAs are a class of small noncoding RNAs that function similar to trans-acting elements, mainly binding to target gene mRNAs in the form of complementary base pairing, mediating degradation of target genes or inhibiting translation, and thus participating in the regulation of target gene expression. miRNAs play an irreplaceable role in plant growth and development. Previous studies have shown that changes in miRNA expression level can lead to non-additive expression of its target genes and affect seed development, including seed embryo development and seed maturation [Citation11]. For example, Zhang et al. [Citation12] overexpressed miR397 in rice and observed increased grain size, spikelet differentiation and 25% increased field yield. On the other hand, siRNA also plays an important role in gene expression regulation [Citation13, Citation14]. It mainly regulates gene expression by mediating genomic DNA methylation, and siRNAs are particularly associated with transposable elements (TEs) [Citation15]. Lu et al. [Citation16] found that 24-nt siRNAs were significantly expressed in A. thaliana seed development and were involved in the expression regulation of transposable element-associated genes (TAGs). Li et al. [Citation17] showed that 33 siRNAs participate in the expression regulation of neighboring TAGs in hexaploid wheat. These studies suggest that siRNAs may be involved in genome-wide regulation of gene expression and are essential for seed development in plants.
Flax (Linum usitatissimum L.) is one of the most important industrial crops in the world [Citation18]. Linseed is rich in a variety of bioactive substances such as α-linolenic acid and lignans [Citation19], which give linseed a variety of effects such as anti-cancer, blood lipid lowering, blood sugar lowering and cardiovascular disease treating effects [Citation20–23]. Developing flaxseed powder with different health care functions will have great economic and social benefits by taking high quality flaxseed as raw material and making full use of its nutritional and disease prevention functions. However, there is little research on seed development of flax. Venglat et al. [Citation24] took the flax variety ‘Bethune’ as research material and used 13 libraries to analyze the seeds of flax at different developmental stages to generate 261,272 expressed sequence tags (ESTs), revealing the gene expression dynamics of many important components of bioscience during seed development. Xie et al. [Citation25] used 224 samples of flax natural population material and combined SLAF-seq sequencing data and genome-wide association study (GWAS) of seed fatty acid content. Low oil Shuangya 4 varieties and new high oil varieties in three different periods of seed development were subjected to transcriptome sequencing (RNA-seq), and analysis of candidate genes to characterize fatty acid metabolism in flaxseed was conducted with two methods.
Zhang et al. [Citation26] used four small RNA libraries from early flax seeds at 5, 10, 20 and 30 DPA were constructed and used for high-throughput sequencing to identify miRNAs. That study preliminarily filled the gaps in identification and functional analysis of miRNA in flax seed development. However, its study did not conduct degradation group validation, nor did it conduct relevant miRNA-target genes analysis. In most cases, miRNA silences gene expression by degrading target gene mRNAs, and it is particularly important to understand the function of miRNA by identifying miRNA-target genes. Based on this, we carried out a comparative analysis of the degradation group and the dynamic changes of miRNA transcription levels at three important stages (10 DPA, 20 DPA and 30 DPA) of flaxseed development. The dynamic changes of miRNA and target gene mRNAs identified in this study can provide an important reference for the analysis of flaxseed development [Citation26].
Materials and methods
Preparation of experimental materials and construction of sequencing library
Shuangya 4 is the main flax cultivar of Heilongjiang Province in China and has strong stress resistance, high seed yield and excellent comprehensive characteristics. It was planted in the Minzhu experimental field of Heilongjiang Academy of Agricultural Sciences (N45°51’, E126°48’) with normal field management. The labels were attached after the flax plants flowered, and samples that included three stages of seed development of flax (namely, 10 DPA, 20 DPA and 30 DPA) were collected from 8:30 to 9:30 every morning. Each sample consisted of three biological replicates. To ensure consistency, each sample consisted of 10–15 uniform seeds. cDNA and small RNA libraries were constructed according to Illumina’s instructions.
Small RNA sequencing and data analysis of flaxseed
The frozen flaxseed samples from three stages of seed development were ground in liquid nitrogen using a mortar and pestle. The total RNA was extracted using the RNA simple Total RNA kit (Tiangen Biotech, Beijing, China), according to the manufacturer’s protocol. The RNA content was calculated using the Bioanalyzer 2100 algorithm (Agilent Technologies, Palo Alto, USA). After the RNA samples were tested to be of satisfactory quality, 1.5 µg was used as the starting amount of RNA samples and the volume was adjusted to 6 µL with water. The library was constructed using the small RNA Sample Pre-Kit (Norgen Biotek, Canada). Since the 5’ end of small RNA has a phosphate group and the 3’ end has a hydroxyl group, the T4 RNA Ligase 1 and T4 RNA Ligase 2 (truncated) were used to connect joints at the 3’ end and 5’ end of small RNA. The first strand of cDNA was synthesized by reverse transcription, and the double-strand library was obtained by polymerase chain reaction (PCR) amplification using cDNA as a template. In the small RNA library, the target fragments were purified by polyacrylamide gel [Citation27]. Qubit 2.0 was used to detect the library concentration. The library concentration was diluted to 1 ng/μL, and the Insert Size was detected by Agilent 2100 bioanalyzer. The effective concentration of the library was accurately quantified by qRT-PCR to check the library quality. HiSeq2500 was used for high-throughput sequencing with a read length of 50 nt for single-end (SE) reads. Silva database, GtRNAdb database, Rfam database and Repbase database were used to align with clean reads. Raw miRNA sequences were deposited in the National Center for Biotechnology Information (NCBI) database and can be accessed in the database (https://www.ncbi.nlm.nih.gov/) under accession number PRJNA478806.
Identification of differentially expressed miRNAs
Statistics were conducted on miRNA expression in each sample. The expression was normalized with the TPM algorithm [28]. In the detection of differentially expressed miRNA, |log2(FC)| > = 1 and FDR < = 0.05 were used as the screening criteria. Fold Change (FC) refers to the ratio of the expression amount between two samples. In the statistical analysis we applied the null-hypothesis. Because the miRNA expression analyses were independent and a large number of statistical hypothesis tests were conducted for miRNA expression, the problem of false positives may occur during the analysis. The Benjamini-Hochberg correction method of hypothesis testing was used to correct the original significance p values (p-value), and finally the False Discovery Rate (False Discovery Rate, FDR) was used as the key indicator of screening differentially expressed miRNAs. The Volcano Plot was used to examine the differences in miRNA expression levels between the two samples, as well as the statistical significance of the differences.
Prediction and functional annotation of differentially expressed miRNA target genes
According to the sequencing information of known miRNAs and new miRNAs, TargetFinder software was used for target gene prediction [Citation29]. To further understand the function of the genes of interest, we performed pathway annotation analysis using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. Differentially expressed miRNA target gene annotations were classified according to the type of KEGG pathway.
Degradome sequencing and data analysis
Flaxseed samples were obtained from Shuangya 4 flax plants at 10 DPA, 20 DPA and 30 DPA, and RNA was extracted from each sample. mRNA was captured using magnetic beads followed by the ligation of 5’ adapters. Reverse transcription was then performed using mRNA templates and biotinylated random primers, followed by PCR amplification to create the degradome libraries. The prepared libraries were then sequenced using an Illumina HiSeq 2500 system.
The adapter sequences and low-quality tags were removed from the raw tags to obtain clean tags and cluster tags (clustered data of clean tags). The cluster tags were compared to the flax reference genome to obtain the distribution of tags within the genome (https://phytozome.jgi.doe.gov/pz/portal.html) [Citation30]. A comparison was then conducted between the cluster tags and the Rfam database to annotate noncoding RNAs. The unannotated sequences were used for cleavage site analysis. The Cleaveland software [Citation31] was used to predict degradation sites (set conditions of P-value < 0.05). The raw degradation group sequences were deposited in the NCBI database and can be accessed in the database (https://www.ncbi.nlm.nih.gov/) under accession number PRJNA478812.
qRT-PCR experimental verification
We used qRT-PCR to verify some differentially expressed genes identified by high-throughput sequencing. Fresh seed samples at five different developmental stages of 0, 5, 10, 20 and 30 DPA were collected. The samples were put into liquid nitrogen immediately and stored at −80 °C until RNA isolation. All quantitative reactions were run on the ABI PRISM 7300 instrument (Applied Biosystems Inc., USA) with SYBR Premix Ex Taq (Takara Bio Inc. Japan). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was the internal reference gene [Citation32]. A pair of primers for each gene were designed using Primer Premier 5.0 software based on the CDS sequences. The target genes’ primers were checked using the BLAST program in the reference genome of Linum usitatissimum v1.0 [Citation30] to ensure unique amplification. miRNA quantification by qRT-PCR was based on stem-loop primers [Citation33]. The sequences of the specific primers are listed in Table S1, supplementary material. The relative expression levels were calculated as 2-ΔΔCT, where ΔΔCT = (CT, target − CT, GAPDH) time x − (CT, target − CT, GAPDH) time0 [Citation34]. All qRT-PCR experiments were performed for three biological replicates with three technical repeats each under the same conditions.
Table 1. Data set summery of 3 seeds at different developmental stages in flax.
Results
Small RNA library sequencing data analysis at different development stages of flaxseed
MiRNA sequencing was performed on seeds of flax (10 DPA, 20 DPA and 30 DPA). There were three samples for each period, and 9 sample libraries were constructed. We took the average of three replicates of seed library data from three development periods of flax for demonstration (). After sequencing, 22,741,778, 19,888,514 and 15,078,983 raw reads were generated from seeds at 10 d, 20 d and 30 d after flax flowering. After removing the connector, contaminated and low-quality reads, the effective clean reads were 21,275,574, 17,929,236 and 13,590,317, accounting for 99.12%, 99.10% and 99.21% of the total reads, respectively.
Clean reads were aligned with the Silva database, GtRNAdb database, Rfam database and Repbase database. Noncoding RNAs such as ribosomal RNA (rRNA), transport RNA (tRNA), small nuclear RNA (snRNA), small nucleolar RNA (snoRNA) and repeat sequences were filtered to obtain Unannotated reads containing miRNA. The results showed that in the three sample libraries, except for rRNA (12.98%–17.88%), the proportion of other noncoding RNAs was relatively low; for example, scRNA and snRNA were the lowest (0.00%), snoRNA was the second lowest (0.01%), and tRNA accounted for 0.71%–1.27%. The unannotated reads were aligned with the reference flax genome by using miRDeep2 software to obtain location information on the reference genome. In the three sample libraries, the number of miRNAs of the total that can be compared was 18,358,146, 14,536,070 and 11,401,719, respectively. The length distribution of the miRNA sequences in different sample databases was further analyzed on the basis of the existing sequence analysis (). In different data libraries, the miRNA distribution length ranged from 18 to 30 nt. Among them, 21–24 nt accounted for the highest proportion. This is the length range of typical Dicer enzyme-induced shear products [Citation35].
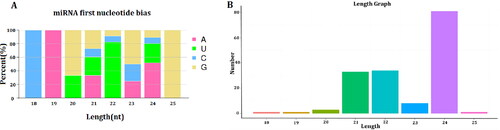
Figure 1. Analysis of length and miRNA first nucleotide bias in novel miRNAs. (A) miRNA first nucleotide bias analysis of novel miRNAs; (B) length analysis of novel miRNAs.
Analysis and identification of known and new miRNAs in flaxseed at different development stages
A total of 130 known miRNAs were identified in the miRNA database of flaxseed in three developmental stages, and they belonged to 29 conserved miRNA families, including the miR156, miR159 and miR160 families. The miR166 family has the most members of the 29 miRNA families, including 14 miR166 genes (Table S2, supplementary material). Some members came from the same precursor gene in different miRNA families and had differences of 1–2 bases. These miRNA genes were isomers of miRNAs and participate in the formation of complex miRNA clusters, but it is not clear whether there were differences in their functions.
Additionally, a total of 93 new miRNAs were identified, belonging to 49 conserved miRNA families. Among them, the miR2118 family had the most members, including 14 miR2118 genes (Table S3, supplementary material). The length of these new miRNAs ranged from 18 to 25, and 24 nt accounted for the highest proportion. Furthermore, the first base preference in the maturation sequence of the new miRNAs was calculated. The preferred first base in the 21 nt and 23 nt length of new miRNAs was ‘G’. The preferred first bases of the new miRNAs with lengths of 22 nt and 24 nt were ‘U’ and ‘A’, respectively (). Additionally, these unmatched miRNA fragments might be flax specific-miRNAs. The precursor sequences were obtained by comparison with the flax genome. Each new miRNA and its corresponding star sequences were basically derived from the same precursor genes.
A total of 235 miRNAs were identified, which included 114 known conserved miRNAs and 121 novel miRNAs [Citation26]. The results of that study differ slightly from ours. We hypothesized that the main reason for this discrepancy was that the two experiments selected different test materials (Macbeth is high in fatty acid content, but Shuangya4 is low in fatty acid content).
Expression patterns of miRNAs in seeds of flax at different developmental stages
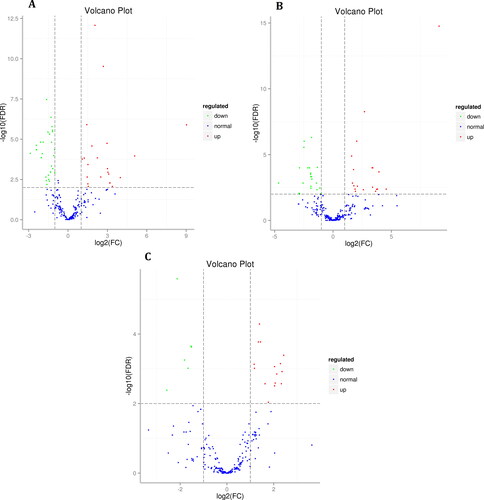
The expression patterns of miRNAs in tissues at different developmental stages can reflect their regulatory functions in growth and development to some extent [Citation36, Citation37]. Therefore, the difference between the miRNA expression levels in each two samples and the statistical significance of the difference can be visualized with a Volcano Plot (). In the 10 d vs. 20 d comparison, there were a total of 36 known differentially expressed miRNAs. Among them, 7 miRNAs were upregulated by |log2FC|>2, and 5 miRNAs were downregulated by |log2FC|>2. A total of 49 new miRNAs were differentially expressed. Among them, 15 miRNAs were upregulated by |log2FC|>2, and 5 miRNAs were downregulated by|log2FC|>2 (; Table S4, supplementary material). In the 10 d vs. 30 d group, a total of 36 known miRNAs were differentially expressed. Among them, 6 miRNAs were upregulated by |log2FC|>2, and 10 miRNAs were downregulated by |log2FC|>2. A total of 42 new miRNAs were differentially expressed. Among them, 15 miRNAs were upregulated by |log2FC|>2, and 8 miRNAs were downregulated by |log2FC|>2 (; Table S5, supplementary material). In the 20 d vs. 30 d comparison group, there were a total of 15 known differentially expressed miRNAs. Among them, 6 miRNAs were downregulated by |log2FC|>2. A total of 20 new miRNAs were differentially expressed. Among them, 8 miRNAs were upregulated by |log2FC|>2 (; Table S6, supplementary material). The above analysis found that the number of differentially expressed miRNAs between the development of flax capsules (0–10 DPA) and the ripening of flax capsules (10–20 DPA) of flaxseed was large. miRNAs that were significantly upregulated included miR171, miR164, miR408 and miR398 family members. miRNAs that were significantly downregulated included miR156 and unconservative_scaffold3872_46150 family members. These miRNAs may play an important regulatory role in the accumulation and growth of flaxseed.
Figure 2. Volcano Plot of miRNAs in developmental stage seeds of flax. The present study used flaxseed of Shuangya 4 at 10, 20 and 30 DPA as the material. (A) 10 d vs. 20 d; (B) 10 d vs. 30 d; (C) 20 d vs. 30 d.
Identified miRNA target genes of flax by the degradation group
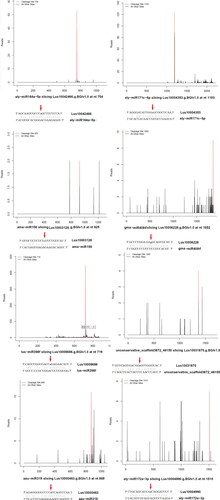
This study used 10 DPA, 20 DPA and 30 DPA seeds of flax as material for high-throughput sequencing to conduct degradation group sequencing to determine the target genes of miRNA in seeds of flax at different development stages. Degradation group sequencing is the most common and efficient analysis method to identify miRNA target genes. We constructed a library of flaxseed degradation groups and conducted in-depth sequencing. MiRNA target genes of flaxseed were identified by combining the previous information of high-throughput sequencing of flaxseed at different developmental stages. Three degradation group cDNA libraries (F01, F02, F03) were constructed from three seeds of Shuangya 4 at different development stages (10 d, 20 d, 30 d). The degradation sites were predicted by Cleaveland software. We found that 82 target genes were degraded, and 82 target gene degradation sites were detected in sample F01; 95 target genes were degraded, and 95 target gene degradation sites were detected in sample F02; 85 target genes were degraded, and 85 target gene degradation sites were detected in sample F03. By integrating the data of 3 degradation groups, 96 target genes of 19 miRNA families were obtained, and 96 degradation sites were detected. Analysis revealed that only 2% of miRNA target genes in flaxseed were identified in the degradation group, and a large number of target genes were still not identified. Through further data analysis and collation, the base complementation and cleavage sites of miRNA and target genes were obtained (Table S7, supplementary material). Part of the t-plot of 96 target genes of 19 miRNA families was plotted figures according to the sequencing results of the degradation group (). The 96 degraded target genes were functionally annotated to explore the function of target genes, and we found that 34 target genes were related to plant growth and development, such as Auxin response factor (lus10005970, lus10007386, lus10010969, lus10016090, lus10017640, lus10017641, lus10019940, lus10020805, lus10021467, lus10023519, lus10024754, lus10026510, lus10030240, lus10031354, lus10033597, lus10033598, lus10040403), SBP domain (lus10012020, lus10016275, lus10023818, lus10036812, lus10003216, lus10040568), AP2/ERP transcription factor (lus10004990, lus10036141, lus10040365, lus10009374, lus10019905, lus10023165, lus10026477), growth regulate factor (lus10011558 and lus10019274) and GRAS family (lus10004353 and lus10028934). Sixteen target genes were annotated as transcription factors, for example, transcription factor NAC (lus10042466, lus10026200, lus10021659, lus10001648), transcription factor bHLH (lus10021968, lus10031255, lus10031826, lus10041261), transcription factor TCP (lus10002195 and lus10023297) and transcription factor MYB (lus10008685 and lus10026142). Seven target genes had annotated function related to plant secondary metabolism, such as Laccase (lus10035517, lus10027782, lus10024378, lus10017175), copper chaperone for superoxide dismutase (lus10006686 and lus10007031), P450 secondary metabolism (lus10014741) and fucosyltransferase family (Lus10006228) (Table S8, supplementary material).
Figure 3. Target plot (t-plot) of target genes of Shuangya 4 identified by degradome sequencing. The arrows are used to annotate the cleavage sites of the target genes.
qRT-PCR verified the regulatory relationship between miRNA-target genes
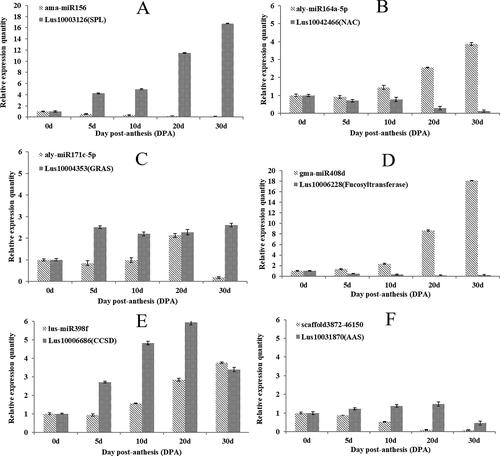
In order to better understand the effects of miRNAs on the target genes of flax seed development, we added two more stages (0d and 5d) in addition to the three stages (10d, 20d and 30d) of seed development selected by transcriptome and degradation group sequencing. Therefore, seed materials of 5 developmental stages (0 d, 5 d, 10 d, 20 d, 30 d) were used in the validation of expression patterns. At the same time, six differentially expressed miRNAs (ama-miR156, aly-miR164-5p, aly-miR171c-5p, gma-miR408d, lus-miR398f and scaffold3872-46150) related to plant growth and secondary metabolite accumulation and their corresponding target genes identified by degradation groups were selected for expression pattern analysis (). MiR156 mainly regulates squamosa promoter binding protein like (SPL) transcription factors [Citation38, Citation39]. The expression level of ama-miR156 decreased steadily with seed development, whereas the expression level of its target gene lus10003126 increased gradually with seed development. The target gene was identified as having SPL function from the degradation data. We speculated that it might be an important target gene of flax miR156 (). However, lus10003126 annotated function in phytozome is Leucine Rich Repeat (LRR). The specific function of this gene should be verified by further experiments. miR164 mainly regulates cup-shaped Cotyledon (CUC) to regulate plant tissue development. The expression pattern of aly-miR164a-5p gradually increased with seed development, and the expression of target gene lus10042466 was basically negatively regulated by aly-miR164a-5p with flaxseed development. This suggests that it may be the main target gene of flax miR164, which is strictly regulated by miR164 to participate in flaxseed development (). miR171 mainly regulates GRAS transcription factors [Citation40, Citation41], and only one flax GRAS target gene, lus10004353, was verified by prediction and degradation group sequencing. There was no obvious relationship between this target gene and the expression pattern of aly-miR171c-5p in different developmental stages of flaxseed, and only a significant negative regulation relationship was found in 30 DPA. miR408 is a highly conserved family in plants, and its conserved target genes include copper ion binding protein, laccase and sugar invertase [Citation42, Citation43]. In this study, the expression level of gma-miR408d increased steadily with the development of seeds. Its target gene lus10006228 was identified as having fucosyltransferase function from the degradation data (Table S8, supplementary material), but it was annotated as k13150-coilin according to phytozome. The gene function needs to be verified by further experiments. It showed the opposite expression pattern of gma-miR408d (except at 30 DPA). This indicates that miR408 may play an important role in seed energy material storage during the mid-early stage of flaxseed development (). miR398 is involved in the regulation of copper homeostasis by reducing the expression of two copper superoxide dismutase genes (CSD). According to , the expression of the target gene increased steadily during 0 d to 20 d of flaxseed development but decreased at 30 d similar to lus-miR398f (). Also, the expression of lus10031870 did not show significant increasing trends, and even dropped down at 30 d. We speculated that scaffold3872-46150 may be involved in the development or substance accumulation of flaxseed, but its specific regulatory function still needs to be further explored (). The results suggested that these miRNAs and target genes interacted with each other and jointly participated in the development and accumulation of secondary metabolites in flax at different stages. We found that the expression patterns of the selected miRNA in each stage of flaxseed development were basically consistent with those in the Small RNA sequencing data (, supplementary material). The above results showed the reliability of the observations in this study.
Figure 4. Expression patterns of each miRNA and their target gene in each stage of flaxseed development. The total RNA of the flax plants was obtained from 0–30 DPA to facilitate analysis of the expression patterns of each miRNA and their target genes in each stage of flaxseed development. GAPDH was used as the internal reference gene for qRT-PCR. The qRT-PCR data are the mean of three biological replicates, whereas the bars represent the standard deviations.
Discussion
Seed development is one of the most important stages in plant life, and it is also a complex biological process. Therefore, numerous studies have been carried out to analyze its potential regulatory mechanism [Citation2, Citation44–46]. Previous studies have shown that the spatiotemporal expression of genes is an important molecular basis for the transformation of plant seeds at different developmental stages [Citation3]. An increasing number of studies have found that some small RNA molecules play an important role in the regulation of gene expression, such as miRNA and siRNA [Citation9, Citation16, Citation47]. Therefore, it was necessary to integrate miRNA and degradation data to provide important information for the analysis of plant seed development. In this study, we integrated miRNA and high-throughput sequencing data from the degradation group. The miRNAs which changed their expression level during flaxseed development were analyzed at the genome-wide level. miRNAs were identified among three important developmental stages of flaxseed. The dynamic changes in miRNA-target gene expression could provide an important reference, and the potential biological functions of miRNAs in the seed’s development were analyzed.
The regulatory pathways of miRNA-target genes play an important role in the growth and development of plants. Some conserved regulatory combinations of ‘miRNA-target genes’ participate in different processes of plant morphogenesis [Citation48]. Among them, some key miRNAs and their target genes play a regulatory role in seed development and were very conserved in different plants; these key miRNAs include miR397-LAC, miR164-ARF and miR156-SPL [Citation49, Citation50]. Seed development includes the development of the embryo and endosperm and the accumulation of seed storage materials. miRNAs regulate seed development through hormone signal transduction, antioxidant action, sugar transformation, cell growth and other action pathways. In our study, miR156, miR171, miR164, miR408, miR398 and miR396 were identified as miRNA families that were differentially expressed during flaxseed development. The expression level of ama-miR156 showed a decreasing trend during the development of flaxseed. miR156 is a class of miRNAs that regulate cell growth. They target SPL10 and SPL11 and cause abnormal cell division in Arabidopsis [Citation51]. It was found that Lus-miRNA156a was significantly correlated with the flax seed development process. An over-expression vector was constructed and transformed to Arabidopsis. The phenotypes of homozygous transgenic lines showed decreasing of oil content and most of the fatty acid content in seeds as well as late flowering time [Citation26]. miR156 targets SPL10/SPL11 to inhibit their expression and control seed development in the early stages of embryonic development in Arabidopsis and rice (10 DPA) [Citation52, Citation53]. This indicated that miR156 inhibits SPL10/SPL11 less in the middle and late stages (20 DPA and 30 DPA) of flaxseed embryo development, to promote normal embryo development. The expression level of miR171 increased in seeds from 10 DPA to 20 DPA, whereas the expression level changed slightly from 20 DPA to 30 DPA. miR171 can negatively regulate some transcription factors encoding the GRAS domain by shearing mRNA in plants [Citation41, Citation54]. The N terminus of the GRAS protein has transcriptional activation activity. It can bind to the promoter of meiosis-related genes in anther cells in the early stage of meiosis and plays a role in transcriptional activation to promote cell division [Citation55]. miR171’s negative regulation of GRAS began to weaken in later stages of flaxseed development, thus promoting cell division and accelerating seed development. The expression level of miR164 increased significantly with the development of flaxseed, and the miR164-mediated regulation of auxin can regulate seed grouting [Citation56]. miR164 is an auxin correlative miRNA which exists in both high quality and low quality panicles of rice and has higher expression in high quality panicles [Citation57]. The expression of miR164 was found to rise in developing wheat seeds, and interference with the regulation of miR164 leads to abnormal embryonic development, indicating that miR164 is involved in seed development through the regulation of auxin signal transduction by its target gene NAC [Citation58]. We speculated that miR164 may play an important role in the nutrient reserves of flaxseed. The expression of miR408 increased gradually during the three stages of flaxseed development. It was found that the expression level of miR408 was positively correlated with the sucrose content in the seed through measuring the content of sugar in the embryos of transgenic rice of the miR408 strain. However, the expression level of its target gene was negatively correlated with the sucrose content of the seed [Citation43]. Thus, miR408 may be involved in the development of plant seeds through the sugar transformation pathway. In this study, the expression of miR398 increased with the development of flaxseed. Some reports have shown that miR397, miR398 and miR528 are highly expressed in rice seed embryos, and their target genes encode copper-binding proteins [Citation59, Citation60]. Sugar-mediated miR398 is involved in copper homeostasis regulation by reducing the expression of two CSD genes [Citation61]. This indicated that the regulatory effect of miR398 was also conserved in flax. Important information was provided by the above reports, such as analysis of differentially expressed miRNAs, screening of candidate miRNAs related to seed development, and identification of relevant regulatory functions of ‘miRNA-target genes’ in flaxseed at different development stages. Scaffold3872-46150 is a novel-miRNA whose function is not yet clear. However, its target gene lus10031870 functions as an asparagine synthase gene. Asakura et al. [Citation62] found that the expression of asparagine synthase gene Oryzasin1 increased 2–4 weeks after flowering in rice. The expression of Oryzasin1 decreased after seed maturation. This suggests that Oryzasin1 may be involved in the processing and degradation of stored proteins, indicating that Oryzasin1 plays an important role in seed formation [Citation62]. In this study, the expression level of lus10031870 gradually increased with the development of flaxseed and began to decline after seed maturity (30 d). This was consistent with the research results of Asakura et al. [Citation62]. Therefore, we speculated that lus10031870 corresponding miRNA scaffold3872-46150 should also have the function to regulate protein accumulation and metabolism of flaxseed and may have an important regulatory role in flaxseed development.
Understanding seed development in different stages of flax could provide important information for flax breeding and germplasm research. miRNA-mediated regulatory mechanisms are important for organ morphological development and cell type differentiation. They are also ideal biological means for the functional study of genes [Citation63, Citation64]. However, there are few reports describing which molecular mechanisms of miRNA-target genes are involved in the regulation of flaxseed development. Screening the regulatory combination of different miRNA-target genes in flaxseed development will provide experimental basis for the future research.
Conclusions
In this study, 130 known miRNAs and 93 new miRNAs were identified. A total of 3,361 target genes were predicted, including 2401 target genes corresponding to known miRNAs and 960 target genes corresponding to new miRNAs. A large number of known miRNAs were differentially expressed in seeds of flax at different development stages, suggesting that they may play an important regulatory role in seed development. Additionally, it was found that known miRNAs including ama-miR156, aly-miR171c-5p, aly-miR164a-5p, gma-miR408d, lus-miR398f and the new miRNA unconservative_scaffold3872_46150 were differentially expressed in the development of flaxseed. These miRNAs and their corresponding target genes were verified by qRT-PCR during the development of flaxseed, and their expression patterns were found to be basically consistent with those described in the sequencing results. This study provides data supporting the functional study of miRNA-target gene regulation.
Author contributions
D.X. and Y.Y. carried out most of the experimental work; Collections of flax germplasm resources were performed by J.S. and Z.D.; D.X. designed the research and wrote the manuscript; J.S. revised the manuscript. All authors read and approved the final manuscript.
Ethics statement
The plant materials were obtained from the Chinese Crop Germplasm Resources System (CGRIS; http://www.cgris.net/). Sampling of plant materials were performed in compliance with institutional, national, and international guidelines. The materials were publicly available for non-commercial purposes. All the experiments undertaken in this study complied with the current laws of China.
| Abbreviations | ||
| miRNA | = | MicroRNA |
| DPA | = | days post anthesis |
| qRT-PCR | = | real-time quantitative PCR |
| siRNAs: | = | small interfering RNAs |
| TEs: | = | transposable elements |
| TAGs: | = | transposable element-associated |
| ESTs: | = | genes expressed sequence tags |
| GWAS: | = | genome-wide association study |
| RNA-seq: | = | transcriptome sequencing |
| FC | = | Fold Change |
| FDR: | = | False Discovery Rate |
| KEGG: | = | Kyoto Encyclopedia of Genes and Genomes |
| GAPDH: | = | Glyceraldehyde-3-phosphate dehydrogenase |
| rRNA | = | ribosomal RNA |
| tRNA: | = | transport RNA |
| snRNA: | = | small nuclear RNA |
| snoRNA: | = | small nucleolar RNA |
| SPL: | = | squamosa promoter binding protein like |
| LRR: | = | Leucine Rich Repeat |
| CUC: | = | cup-shaped Cotyledon |
| GRAS: | = | goldfish recirculating aquaculture system |
| CSD: | = | copper superoxide dismutase genes |
Acknowledgements
We would like to thanks the National Bast Fiber Crops Germplasm Improvement Center of Flax Branch Center for kindly supplying the experimental platform.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Disclosure statement
The authors declare no conflict of interest.
Supplemental material
Raw Illumina sequences were deposited in the National Center for Biotechnology Information (NCBI) and can be accessed in the database (http://www.ncbi.nlm.nih.gov/biosample/7551536) under accession PRJNA478806 and PRJNA478812.
Additional information
Funding
Funding
References
- Tzafrir I, Pena-Muralla R, Dickerman A, et al. Identification of genes required for embryo development in Arabidopsis. Plant Physiol. 2004;135(3):1206–1220.
- Druka A, Muehlbauer G, Druka I, et al. An atlas of gene expression from seed to seed through barley development. Funct Integr Genomics. 2006;6(3):202–211.
- Pfeifer M, Kugler KG, Sandve SR, International Wheat Genome Sequencing Consortium, et al. Genome interplay in the grain transcriptome of hexaploid bread wheat. Science. 2014;345(6194):1250091.
- Sreenivasulu N, Altschmied L, Radchuk V, et al. Transcript profiles and deduced changes of metabolic pathways in maternal and filial tissues of developing barley grains. Plant J. 2004;37(4):539–553.
- Sreenivasulu N, Usadel B, Winter A, et al. Barley grain maturation and germination: metabolic pathway and regulatory network commonalities and differences highlighted by new Map Man/Page Man profiling tools. Plant Physiol. 2008;146(4):1738–1758.
- Wang L, Xie W, Chen Y, et al. A dynamic gene expression atlas covering the entire life cycle of rice. Plant J. 2010;61(5):752–766.
- Devic M. The importance of being essential: EMBRYO-DEFECTIVE genes in Arabidopsis. C R Biol. 2008;331(10):726–736.
- Jones-Rhoades MW, Bartel DP, Bartel B. MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol. 2006;57(1):19–53.
- Curaba J, Spriggs A, Taylor J, et al. miRNA regulation in the early development of barley seed. BMC Plant Biol. 2012;12:120.
- Li PY, Wang H, Liu L, et al. Physiological and transcriptome analyses reveal short-term responses and formation of memory under drought stress in rice. Front. Genet. 2019;10(55):1–16.
- Heisel SE, Zhang Y, Allen E, et al. Characterization of unique small RNA populations from rice grain. PloS One. 2008;3(8):e2871.
- Zhang YC, Yu Y, Wang CY, Li ZY, et al. Overexpression of microRNA OsmiR397 improves rice yield by increasing grain size and promoting panicle branching. Nat Biotechnol. 2013;31(9):848–852.
- Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136(4):642–655.
- Fei Q, Xia R, Meyers BC. Phased, secondary, small interfering RNAs in posttranscriptional regulatory networks. Plant Cell. 2013;25(7):2400–2415.
- Matsunaga W, Ohama N, Tanabe N, et al. A small RNA mediated regulation of a stress-activated retrotransposon and the tissue specific transposition during the reproductive period in Arabidopsis. Front Plant Sci. 2015;6(48):1–12.
- Lu J, Zhang CQ, Baulcombe DC, et al. Maternal siRNAs as regulators of parental genome imbalance and gene expression in endosperm of Arabidopsis seeds. Proc Natl Acad Sci U S A. 2012;109(14):5529–5534.
- Li AL, Liu DC, Wu J, et al. mRNA and small RNA transcriptomes reveal insights into dynamic homoeolog regulation of allopolyploid heterosis in nascent hexaploid wheat. Plant Cell. 2014;26(5):1878–1900.
- Vaisey-Genser M, Morris DH. Introduction: history of the cultivation and uses of flaxseed. In: Muir AD, Westcott ND, editors. Flax: The Genus Linum. Boca Raton (FL): CRC Press; 2003. p. 1–21.
- Wisker E, Rabe E, Metzner C, et al. The effectiveness of linseed[J]. Ernhrungs Umschau. 1999;46(3):76–86.
- Alhassane T, Xu XM. Flaxseed lignans: source, biosynthesis, metabolism, antioxidant activity, bio-active components, and health benefits. Compr Rev Food Sci. 2010;9:261–269.
- Zanwar AA, Hegde MV, Bodhankar SL. Chapter 71—Flax lignan in the prevention of atherosclerotic cardiovascular diseases. In: Polyphenols in human health & disease. Academic Press, Elsevier Inc; 2014. p. 915–921.
- Kreydin EI, Kim MM, Barrisford GW, et al. Urinary lignans are associated with decreased incontinence in postmenopausal women. Urology. 2015;86(4):716–720.
- Chen FP, Chang CJ, Chao AS, et al. Efficacy of Femarelle for the treatment of climacteric syndrome in postmenopausal women: an open label trial. Taiwan J Obstet Gyne. 2016;55(3):336–340.
- Venglat P, Xiang DQ, Qiu SQ, et al. Gene expression analysis of flax seed development. BMC Plant Biol. 2011;11(1):74.
- Xie DW, Dai ZG, Yang ZM, et al. Combined genome-wide association analysis and transcriptome sequencing to identify candidate genes for flax seed fatty acid metabolism. Plant Sci. 2019;286:98–107.
- Zhang TB, Li Z, Song XX, et al. Identification and characterization of microRNAs in the developing seed of linseed flax (Linum usitatissimum L.). IJMS. 2020;21(8):2708.
- Citartan M, Tan SC, Tang TH. A rapid and cost effective method in purifying small RNA. World J Microbiol Biotechnol. 2012;28(1):105–111.
- Fahlgren N, Howell MD, Kasschau KD, et al. High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One. 2007;2(2):e219.
- Allen E, Xie Z, Gustafson AM, et al. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell. 2005;121(2):207–221.
- Wang Z, Hobson N, Galindo L, et al. The genome of flax (Linum usitatissimum. L) assembled de novo from short shotgun sequence reads. Plant J. 2012;72(3):461–473.
- Addo-Quaye C, Miller W, Axtell MJ. CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics. 2009;25(1):130–131.
- Huis R, Hawkins S, Neutelings G. Selection of reference genes for quantitative gene expression normalization in flax (Linum usitatissimum L.). BMC Plant Biol. 2010;10:71.
- Hurley J, Roberts D, Bond A, et al. Stem-loop RT-qPCR for microRNA expression profiling. Methods Mol Biol. 2012;822:33–52.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25(4):402–408.
- Kandasamy SK, Fukunaga R. Phosphate-binding pocket in Dicer-2 PAZ domain for high-fidelity siRNA production. Proc Natl Acad Sci U S A. 2016;113(49):14031–14036.
- Parizotto EA, Dunoyer P, Rahm N, et al. In vivo investigation of the transcription, processing, endonucleolytic activity, and functional relevance of the spatial distribution of a plant miRNA. Genes Dev. 2004;18(18):2237–2242.
- Megraw M, Baev V, Rusinov V, et al. MicroRNA promoter element discovery in Arabidopsis. RNA. 2006;12:1612–1619.
- Cui LG, Shan JX, Shi M, et al. The miR156-SPL9-DFR pathway coordinates the relationship between development and abiotic stress tolerance in plants. Plant J. 2014;80(6):1108–1117.
- Wang JW, Schwab R, Czech B, et al. Dual effects of miR156-Targeted SPL genes and CYP78A5/KLUH on plastochron length and organ size in Arabidopsis thaliana. Plant Cell. 2008;20(5):1231–1243.
- Huang W, Peng SY, Xian ZQ, et al. Overexpression of a tomato miR171 target gene SlGRAS24 impacts multiple agronomical traits via regulating gibberellin and auxin homeostasis. Plant Biotechnol J. 2017;15(4):472–478.
- Hai BZ, Qiu ZB, He YY, Yuan MM, et al. Characterization and primary functional analysis of Pinus densata miR171. Biologia Plant. 2018;62(2):318–324.
- Song ZQ, Zhang LF, Wang YL, et al. Constitutive expression of miR408 improves biomass and seed yield in Arabidopsis. Front Plant Sci. 2018;8:2114.
- Ma SY. Expression pattern and function of miR408 in seed development of rice (Oryza sativa). Master’s thesis in China, with English abstract, Zhejiang University, 2012.
- Radchuk VV, Borisjuk L, Sreenivasulu N, et al. Spatiotemporal profiling of starch biosynthesis and degradation in the developing barley grain. Plant Physiol. 2009;150(1):190–204.
- Martinez-Barajas E, Delatte T, Schluepmann H, et al. Wheat grain development is characterized by remarkable trehalose 6-phosphate accumulation pregrain filling: tissue distribution and relationship to SNF1-related protein kinase1 activity. Plant Physiol. 2011;156(1):373–381.
- Haberer G, Mayer KF. Barley: from brittle to stable harvest. Cell. 2015;162(3):469–471.
- Zhou J, Cheng Y, Yin M, et al. Identification of novel miRNAs and miRNA expression profiling in wheat hybrid necrosis. PloS One . 2015;10(2):e0117507.
- Rubio-Somoza I, Weigel D. MicroRNA networks and developmental plasticity in plants. Trends Plant Sci. 2011;16(5):258–264.
- Zhou M, Gu L, Li P, et al. Degradome sequencing reveals endogenous small RNA targets in rice (Oryza sativa L. ssp. indica). Front Biol. 2010;5(1):67–e90.
- Damodharan S, Corem S, Gupta SK, et al. Tuning of SlARF10A dosage by sly-miR160a is critical for auxin-mediated compound leaf and flower development. Plant J. 2018;96(4):855–868.
- Nodine MD, Bartel DP. MicroRNAs prevent precocious gene expression and enable pattern formation during plant embryogenesis. Genes Dev. 2010;24(23):2678–2692.
- Palatnik JF, Allen E, Wu X, et al. Control of leaf morphogenesis by microRNAs. Nature. 2003;425(6955):257–263.
- Wang SK, Wu K, Yuan QB, et al. Control of grain size, shape and quality by OsSPL16 in rice. Nat Genet. 2012;44(8):950–954.
- Wang L, Mai Y-X, Zhang Y-C, et al. MicroRNA171c-targeted SCL6-II, SCL6-III, and SCL6-IV genes regulate shoot branching in Arabidopsis. Mol Plant. 2010;3(5):794–806.,
- Fan S, Zhang D, Gao C, et al. Identification, classification, and expression analysis of GRAS gene family in Malus domestica. Front Physiol. 2017;8:253.
- Peng T, Sun HZ, Qiao MM, et al. Differentially expressed microRNA cohorts in seed development may contribute to poor grain filling of inferior spikelets in rice. BMC Plant Biol. 2014;14:196.
- Yi R, Zhu ZX, Hu JH, et al. Identification and expression analysis of microRNAs at the grain filling stage in rice(Oryza sativa L.)via deep sequencing. PLoS One. 2013;8(3):e57863.
- Han R, Jian C, Lv JY, et al. Identification and characterization of microRNAs in the flag leaf and developing seed of wheat (Triticum aestivum L.). BMC Genomics. 2014;15:289.
- Sunkar R, Zhu JK. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell. 2004;16(8):2001–2019.
- Liu B, Li PC, Li X, et al. Loss of function of OsDCL1 affects microRNA accumulation and causes developmental defects in rice. Plant Physiol. 2005;139(1):296–305.
- Song QX, Liu YF, Hu XY, et al. Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biol. 2011;11:5.
- Asakura T, Watanabe H, Abe K, et al. Rice aspartic proteinase, oryzasin, expressed during seed ripening and germination, has a gene organization distinct from those of animal and microbial aspartic proteinases. Eur J Biochem. 1995;232(1):77–83.
- Luo Y, Guo Z, Li L. Evolutionary conservation of microRNA regulatory programs in plant flower development. Dev Biol. 2013;380(2):133–144.
- Zhang B, Wang Q. MicroRNA-based biotechnology for plant improvement. J Cell Physiol. 2015;230(1):1–15.