
Abstract
Diclofenac is one of the most prescribed non-steroidal anti-inflammatory drugs (NSAIDs) for acute and chronic inflammatory conditions. Taking into consideration the extensive use of diclofenac, the crucial role of inflammation in tumorigenesis and data on the effects of NSAIDs on cancer cell lines, we investigated the cytotoxic potential of diclofenac on a panel of human cell lines originating from breast (MCF-7), cervical (HeLa) and colorectal cancer (HT-29). The cytotoxicity was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) dye reduction assay and the half maximal inhibitory concentrations (IC50) were determined. Diclofenac had higher potency against MCF-7 and HT-29 than against HeLa. The cellular and nuclear changes were examined by fluorescent microscopy using 4′,6′-diamino-2-phenylindole (DAPI) stain and acridine orange/ethidium bromide (AO/EB). The wound-healing scratch assay showed significant reduction in the migration capacity of all tested cancer cell lines. The observed antineoplastic activity implies that the anticancer potential of NSAIDs and diclofenac, in particular, necessitates further investigation.
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are among the most widely used drugs for the treatment of pain, fever and inflammation. NSAIDs sales exceeded 8.5 bln Euros for the period 2016 to 2019, in Europe. In Bulgaria, NSAIDs sales increased from 25 mln Euros, in 2016, to almost 40 mln Euros in 2019. Worldwide, diclofenac, in all pharmaceutical forms, had 95 mln Euros of sales for the three years from 2016 to 2019 [Citation1]. Diclofenac was first introduced in Japan, in 1974, as a sodium salt. It is a commonly prescribed NSAID largely due to its pronounced anti-inflammatory activity [Citation2–4].
It has been documented that NSAIDs suppress tumorigenesis and cancer progression by virtue of different cellular mechanisms [Citation5–7]. Their principal mode of action remained elusive until the 1970s, when John Vane discovered that NSAIDs inhibit cyclooxygenase (COX) and this mechanism of action has been widely accepted [Citation8]. Other putative theories involve inhibition of leukotriene synthesis, inhibition of phospholipase A2, and modulation of arachidonic acid levels [Citation9]. Emerging mechanisms of action, alternative to the COX route, is the modulation of nuclear receptor activity, in particular the peroxisome proliferator activated receptor gamma (PPARγ). PPARγ is involved in fatty acid metabolism, controling differentiation of adipocytes and macrophages. It is also involved in inflammatory processes and has been shown to play a role in suppressing tumour cell proliferation [Citation10]. The role of COX, in tumour progression, has also been extensively studied [Citation11–14]. Diaz-Rodriguez et al. [Citation15] found significant decreases in proliferation versus untreated MG-63 cells after 24 h of incubation with 1 and 10 µmol/L of diclofenac (p < 0.001). Gottfried et al. [Citation16], showed that diclofenac significantly diminished the expression of MYC (a family of regulator genes and protooncogenes that code for transcription factors) and modulated glucose metabolism resulting in impaired melanoma, leukemia and carcinoma cell line proliferation in vitro. Hixson et al. [Citation17] estimated the IC50 values of 18 different NSAIDs (incl. diclofenac), for human colon cancer cells, – HT-29, SW480 and DLD-1. Diclofenac had the lowest IC50 of 55 µmol/L, for the HT-29 cells, meaning that it had high potency against this cell line. Similar results were obtained for the DLD-1 cells. However, IC50 for SW480 was significantly higher (170 µmol/L) [Citation18]. Similar results of different potency, based on the cell line, were observed also in the study of Al-Nimer et al. [Citation19], where diclofenac has a superior effect compared to aspirin against fibroblast, HeLa and rhabdomyosarcoma cell lines growth. The study also showed no significant effect against the growth of mammary adenocarcinoma cell line (AMN3). The results from the aforementioned studies indicate that NSAIDs (incl. diclofenac) have a broad spectrum and pleiotropic antineoplastic effects, which outline the necessity for their further pharmacological evaluation as potential anticancer agents.
The aim of the present study was to examine the cytotoxic and antiproliferative effects of diclofenac on human tumour cell lines representative of some of the most common types of human malignances.
Materials and methods
Materials
Diclofenac sodium (Almiral® solution for injection − 25 mg/mL, Batch No.A206F) was supplied by a local pharmacy to test the actual product available on the market that patients take, not the pure substance. The solutions were diluted ex tempore. Acridine orange (Cat. N. A 6014), ethidium bromide (Cat. N. 46065) and penicillin–streptomycin (P 4333-100 mL) were purchased from Sigma-Aldrich Chemie GmbH (Germany). Foetal bovine serum (FBS) was supplied by Gibco (Austria). Dulbecco’s Modified Eagle’s Medium with 4.5 g/L glucose, without L-glutamine, was purchased from Sigma-Aldrich Chemie GmbH (Germany) and DAPI (4′,6-diamidino-2-phenylindole) was supplied from AppliChem (Germany).
Cell lines
The study was carried out in a panel of human cancer cell lines of different cell type and origin. HeLa human cervical cancer cells, HT-29 human colorectal adenocarcinoma cells and МСF-7 human breast adenocarcinoma cells were obtained from the American Type Cultures Collection (ATCC). The cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) containing penicillin (100 units/mL), streptomycin 100 µg/mL, 2 mmol/L L-glutamine and 10% FBS in T-75 cm2 culture flasks at 37 °C in an incubator with 5% CO2 supply and 95% humidity (CO2–incubator, model 3111; Thermo Fisher Scientific, USA).
Cell viability (MTT) assay
The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) dye reduction assay described by Mosmann [Citation20] was used to assess the cytotoxic effects of diclofenac on HeLa, HT-29 and МСF-7 tumour cell lines. The test is based on the reduction of the yellow tetrazolium salt MTT to purple formazan by the mitochondrial dehydrogenases of viable cells. The tumour cells were plated at a density of 1 × 105cells per well in polystyrene, flat-bottom 96-well microtiter plates in DMEM containing 10% FBS and allowed to adhere overnight in a CO2 incubator at 37 °C. At the end of the incubation, the cells were treated with different concentrations of diclofenac (400 µg/mL, 200 µg/mL, 100 µg/mL, 50 µg/mL, 25 µg/mL, 12.5 µg/mL, 6.25 µg/mL and 3.125 µg/mL) in a final volume of 100 μL/well in triplicate wells for each treatment for 24 h or 48 h at 37 °C in a 5% CO2 incubator. Control cells were grown in DMEM containing 10% FBS without treatment. At the end of the treatment, the culture medium was removed and 100 μL of MTT solution (0.5 mg/mL) was added to each well. The plates were incubated for an additional 3 h in a CO2 incubator. The formazan crystals were dissolved after the addition of 100 μL/well of lysis solution (DMSO: Ethanol = 1: 1). The amount of the formed formazan was measured spectrophotometrically at 540 nm and 620 nm (as a referent) wavelength by an enzyme-linked immunosorbent assay (ELISA) spectrophotometer (TECAN, SunriseTM, Grödig/Salzburg, Austria) and the cell vitality was calculated using the following formula: CV (%) = [OD540 (test)/OD540 (control)] × 100.
Fluorescent microscopy detection of apoptosis
The apoptotic changes in the tumour cells cultured in the presences of diclofenac were studied after double intravital staining with dual acridine orange/ethidium bromide (AO/EB) fluorescent staining and staining with the fluorescent dye 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI) as described by Nikolova et al. [Citation21]. Tumour cells at a concentration of 1 × 105 cells/mL in DMEM medium with 10% FBS were seeded on glass coverslips placed on the bottom of 24-well plates and cultured for 24 h in a CO2 incubator to form a cell monolayer. The next day, the cells were treated with diclofenac at a concentration aproximating the IC50 values for 24 h determined by the MTT test for the respective tumour line (for HeLa, 300 µg/mL; HT-29, 100 µg/mL; MCF-7, 300 µg/mL). The plates were cultured in a CO2 incubator under standard conditions for 24 h. Tumour cells from each line cultured only in medium on glass coverslips were used as controls. After incubation, the glass coverslips were washed twice with phosphate buffered saline (PBS) to remove unadhered tumour cells and were stained with fluorescent dyes.
Double staining with acridine orange and ethidium bromide
Acridine orange (AO) and ethidium bromide (EtBr) (live/dead) staining was performed as previously described [Citation21]. Briefly, cell preparations were stained with the fluorescent dyes AO (5 µg/mL) and EtBr (5 μg/mL) in PBS and mounted on microscope slides. The samples were observed under a fluorescence microscope (Leica DM 5000B, Wetzlar, Germany).
DAPI staining
The alterations in the nuclear morphology of the tumour cells induced by diclofenac were studied after staining with the DNA-binding dye DAPI. The cells were fixed with methanol, incubated for 15 min in 1 µg/mL DAPI in methanol in the dark and were mounted with glycerol on microscope slides. Stained cells were visualized and examined under a fluorescence microscope (Leica DM 5000B, Wetzlar, Germany).
Wound-healing assay
The effect of diclofenac on the migration capacity of HeLa, HT-29 and МСF-7 cell lines was examined by the wound-healing test, which analyses the filling of a wound (created mechanically by making a scratch) in the cell monolayer [Citation22]. Human tumour cells at a concentration of 2.5 × 105 cells/mL (1 mL/well) in DMEM medium containing 10% FBS were seeded in 24 well plates and cultured in a CO2 incubator (at 5% CO2, 37 °C and 95% humidity) for monolayer formation. Then, 24 h later, a vertical cut was created in the monolayer of each well of the plate using a yellow pipette tip (volume 100 µL), the wells were washed with FBS-free DMEM medium to remove scratched cells and were treated with diclofenac at concentrations approximating ½ of the IC50 values established by the MTT test after 48 h treatment (HeLa, 90 µg/mL; HT-29, 40 µg/mL; MCF-7, 25 µg/mL). The plates were cultured for 72 h in a CO2 incubator. Images were taken using an Olympus inverted light microscope with a digital camera at the beginning of the experiment and at regular time intervals (24, 48 and 72 h) to quantify the area of cell migration. Cell monolayers of the corresponding cell type with vertical wounds, cultured only in medium served as controls. The measurement of the area of the wound was performed with the software package ImageJ.
Statistical analysis
The statistical significance of the results was assessed by analysis of variance (ANOVA), followed by post hoc comparison test (Bonferroni) with the use of GraphPAD PRISM software, version 5 (GraphPad Software Inc., San Diego, USA). The data are presented as mean values with standard deviation (±SD). Probability values of р <0.05 were considered statistically significant.
Results
Evaluation of the cell viability by MTT test
The antiproliferative effect of diclofenac on HeLa, HT-29 and MCF-7 human cancer cells was evaluated on the 24th and 48th hour after treatment with eight concentrations (400 μg/mL, 200 μg/mL, 100 μg/mL, 50 μg/mL, 25 μg/mL, 12.5 μg/mL, 6.25 μg/mL and 3.13 μg/mL) ().
Figure 1. Cytotoxic effect of different concentrations of diclofenac on cancer cell lines MCF-7, HeLa and HT-29, determined on the 24th (A) and 48th (B) hour by MTT assay. Values represent mean ± SD.
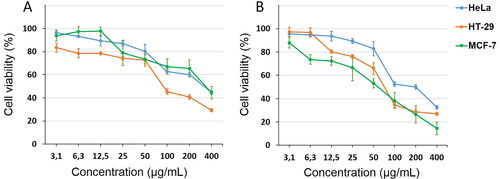
Diclofenac induced a statistically significant decrease in cell viability of HT-29 tumour cells, at all concentrations on the 24th hour (with values from 83.47 ± 5.91 to 29.32 ± 1.31), while the other two cell lines were less sensitive (MCF-7 > HeLa tumour cells). On the 48th hour, the MCF- 7 cell line proved to be the most sensitive at all studied concentrations, followed by HeLa and HT-29. The IC50 values of diclofenac on the 24th and 48th hour were as follows: MCF-7, 348.18 μg/mL (1095 µmol/L) and 46.45 μg/mL (150 µmol/L); HeLa, 313.35 μg/mL (985 µmol/L) and 174.39 μg/mL (548 µmol/L); and HT-29, 111.29 μg/mL (350 µmol/L) and 78.97 μg/mL (248 µmol/L), respectively.
Fluorescent microscopy detection of apoptosis
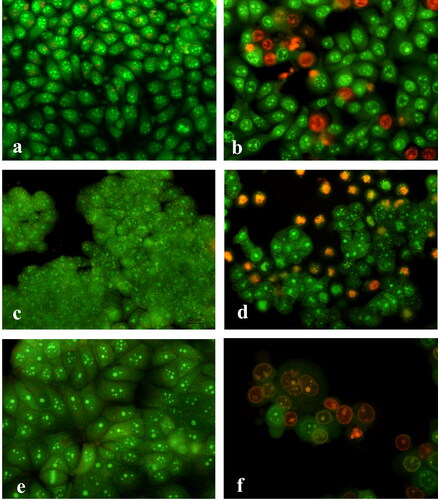
The apoptosis inducing ability of diclofenac was analized by fluorescent microscopy after staining with acridine orange/ethidium bromide and DAPI. The exposure intensity, in this experiment, was based on approximation of the pre-established IC50, for diclofenac, measured at the 24th hour by the MTT assay for each tumour cell line: 300 μg/mL for HeLa; 100 μg/mL for HT-29 and 300 μg/mL for MCF-7. The staining with AO/EtBr showed that after diclofenac treatment the appearance of the tumour cells was modified and morphologic alterations were observed. Cells with bright green nuclei with chromatin condensation in the form of dense green areas (early apoptotic cells) and cells with orange nuclei exhibiting chromatin condensation (late apoptotic cells), nucleus fragmentation and apoptotic body formation, as an indication for cell death induced by the pathway of apoptosis, were observed. The untreated control cancer cell lines had normal growth for the corresponding cell type and retained their distinct morphological features (). HeLa cells treated with diclofenac were rounded in shape, some of them were blistered, with a bright green nucleus, with a condensation of the chromatin in the form of dense green areas (early apoptotic cells). Others were with an orange-colored nucleus, with chroman condensation, and nuclear fragmentation (late apoptotic cells) (). Diclofenac exposure resulted in severe alterations in HT-29 and MCF-7 tumour cell cultures, such as impaired monolayer and three-dimensional growth, reduced cell counts in HT-29 and cell rounding in MCF-7, as well as, presence of cells with morphological alterations typical of apoptosis, like cell membrane blebbing, chromatin condensation, nuclear fragmentation and apoptotic bodies (late apoptosis) as well as necrotic cells ().
Figure 2. Fluorescence microscopy of acridine orange/ethidium bromid stained human tumour cells after 24-h treatment with diclofenac. HeLa cells (a, b): control (a) and treated (b) with 300 µg/mL diclofenac; HT-29 cells (c, d): control (c) and treated (d) with 100 µg/mL diclofenac; MCF-7 cells (e, f): control (e) and treated (f) with 300 µg/mL diclofenac. 40Х magnification.
The DAPI staining revealed that the control-untreated, HeLa, HT-29 and MCF-7 tumour cells had intact nuclei, approximately uniform in shape and size, with smooth edges and homogeneously distributed chromatin (). Cells from the MCF-7 tumour cell line had one to several nucleoli in the nucleus. There were also nuclei of cells in the phase of mitosis ().
Figure 3. Fluorescence microscopy of DAPI stained human tumour cells after 24-h treatment with diclofenac. HeLa cells (a, b): control (a) and treated (b) with 300 µg/mL diclofenac; HT-29 cells (c, d): control (c) and treated (d) with 100 µg/mL diclofenac; MCF-7 cells (e, f): control (e) and treated (f) with 300 µg/mL diclofenac. 40Х magnification.
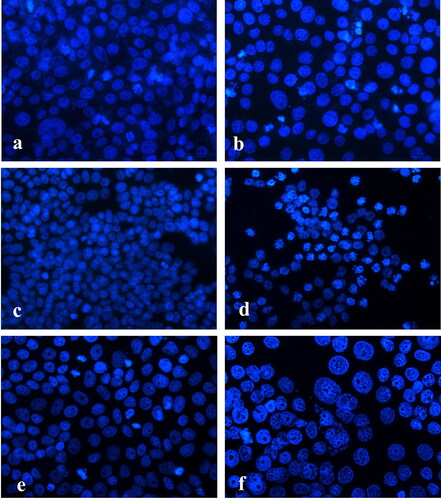
Treatment with diclofenac caused changes in the nuclei of HeLa cells expressed in chromatin condensation (non-homogeneously distributed chromatin with grain structure), fragmented nuclei and formation of apoptotic bodies (). Reduction in cell count, different sized nuclei, uneven edges, unevenly coloured, highly condensed chromatin, apoptotic body formation and nuclear fragmentation (), were observed in the treated HT-29 tumour cells. Treatment in MCF-7 tumour cells, with diclofenac, resulted in extremely severe changes: enlarged nuclei with irregular edges, strong condensation and margination of chromatin, nuclear fragmentation and a number of apoptotic bodies ().
Wound healing assay (scratch assay)
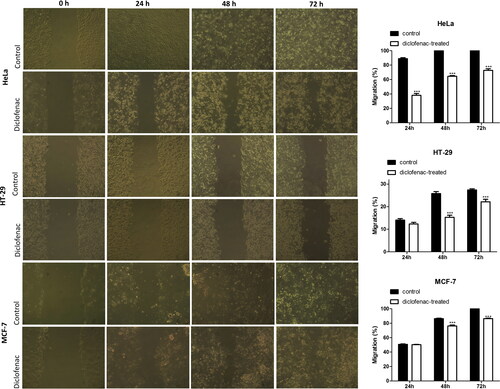
The effects of diclofenac on the migration capacity to the HeLa, HT-29 and MCF-7 tumour cell lines was examined by the scratch wound healing assay. The treatment of HeLa tumour cells with diclofenac showed a time-dependent and statistically significant delay in the healing of the wound at the three study time intervals () compared to the respective untreated control. The most pronounced decrease was found on the 24th hour: 38.15 ± 4.83% compared to the control 88.92 ± 2.69%. At 48 h and 72 h, the migratory capacity of the treated HeLa cells was 64.52 ± 1.8% and 72.75 ± 3.8% respectively, whereas in control cultures the wound was completely healed. Diclofenac did not affect the migration of HT-29 and MCF-7 tumour cells at 24 h. However, at 48 and 72 h, there was a statistically significant inhibition of the migration of HT-29 cells (15.30 ± 1.86% and 22.16 ± 2.25%, respectively for diclofenac-treated cells compared to 25.81 ± 1.79% and 27.43 ± 1.01% for the controls) and the migration of MCF-7 tumour cells (76.24 ± 2.85% and 86.43 ± 1.29%, respectively for diclofenac-treated cells compared to 86.49 ± 1.291% and 100% for the controls).
Figure 4. Effect of diclofenac on the migration of HeLa, HT-29 and MCF-7 human tumour cells. Diclofenac was administered in a concentration equal to 1/2 of the IC50 measured at 48 h by the MTT assay for the respective tumour cell type: HeLa, 90 μg/mL; HT-29, 40 μg/mL; MCF-7, 25 μg/mL.
Discussion
Multiple drug resistance and side effects of conventional antitumor therapy are factors that draw attention to the study of the antitumor potential of drugs established in clinical practice for other applications. These investigations would allow the redirection of old drugs to new applications in practice. In this regard, a series of studies have shown that diclofenac (a commonly prescribed NSAID with analgesic, anti-inflammatory and antipyretic properties, broadly used in treating a variety of acute and chronic pain and inflammatory conditions) inhibited the growth of various cell lines in vitro and induced apoptosis [Citation15–19,Citation23,Citation24]. Diclofenac at a concentration of 0.4 mmol/L significantly suppressed the proliferation of the human melanoma (MelIm) and myeloid leukemia (U937) cell line in vitro. The number of viable U937 cells was reduced to 45% and 31% for 0.1 mmol/L and 0.2 mmol/L concentration, respectively. In contrast, diclofenac at the same concentrations did not impair the viability of normal blood monocytes [Citation16]. Diclofenac treatment at a concentration of 50 μg/mL significantly reduced the proliferation (p < 0.01) of HepG2 hepatocellular carcinoma (HCC) cell line [Citation25]. Our results showed that diclofenac, at all the concentrations studied here, decreased the proliferation of the three tumour cell lines, and this inhibition was dose-dependent. After 24 h of treatment, the inhibitory effect was strongest in HT-29 (colorectal cancer) and almost the same in the other two cell lines (HeLa and MCF-7). The lowest cell viability was 29.32 ± 1.31% in HT-29 at 400 μg/mL of diclofenac. At the 48th hour of the study, the strongest inhibition was found in MCF-7 cells, with values reaching 14.57 ± 4.89% cell viability at a concentration of 400 μg/mL.
A previous study demonstrated that the in vitro anti-proliferative effect of diclofenac against a panel of human glioblastoma cell lines (HTZ-349, U87MG and A172) was observed in a concentrations ranging from 50 μmol/L to 200 μmol/L, which are physiologically relevant [Citation26]. Similarly, concentrations of diclofenac below 200 μmol/L impaired cell growth, and concentrations above 300 μmol/L caused cell death of cultured GL261 glioma cells [Citation27]. In another study, diclofenac (in the range of 20–200 μmol/L) showed the strongest activity, inducing apoptosis and inhibiting cell growth in four ovarian cancer cell lines (SKOV-3, CAOV-3, SW626 and 36M2) [Citation28]. The significantly (p < 0.05) reduced cell viability of ovarian cancer cell lines at concentrations of 50 μmol/L in HEY and OVCAR5 cells, and at 250 μmol/L in the UCl-101 line after 24 h of diclofenac treatment was also reported [Citation29]. In three human colon cancer cell lines (HT-29, SW480 and DLD-1) in vitro anti-proliferative effect of diclofenac was established with IC50 of 55 μmol/L, 37 μmol/L and 170 μmol/L, respectively [Citation17]. The IC50 values of diclofenac for HT-29 cells established in the present study at the 24th and 48th hour were 111.29 μg/mL (350 µmol/L) and 78.97 μg/mL (248 µmol/L), respectively. The higher IC50 values for the HT-29 cell line in our study could be explained with the shorter time of exposure to diclofenac and the different methods for assessment of cell viability used.
A mechanism that could contribute to the anti-proliferative effect of the NSAIDs compounds is apoptosis. Apoptosis, or programmed cell death, is generally characterized by distinct morphological characteristics and biochemical alterations that prevent cancer growth and proliferation [Citation30]. Research has shown apoptosis due to NSAID treatment, including diclofenac, in a panel of ovarian cancer cell lines [Citation28]. Different changes characteristic of early or late apoptosis, in cell and nuclear morphology, were observed in diclofenac-treated hepatocytes, gastric cells, kidney cells and leukocytes [Citation31–34]. A report showed that diclofenac induced growth inhibition and apoptosis of promyelocytic leukemia cells (HL-60) through modulation of mitochondrial functions regulated by reactive oxygen species (ROS), Akt, caspase-8 and Bid [Citation35]. The selective apoptotic action by diclofenac was demonstrated for two human melanoma cell lines, A2058 and SAN, whereas no significant effects were observed in the human non-malignant fibroblast cell line, BJ-5ta [Citation36]. The data obtained by us by staining with acridine orange/ethidium bromide and DAPI also revealed apoptotic changes in the cell and nuclear morphology of diclofenac-treated HeLa, HT-29 and MCF-7 tumour cells. The cell membrane blebbing, chromatin condensation and margination, nuclear fragmentation and formation of apoptotic bodies were the most distinct apoptotic features observed in the tumour cells after the exposure to diclofenac.
Cancer pathogenesis is characterized by rapid tumour cell proliferation and migration. Leidgens et al. [Citation26] investigated the influence of NSAIDs drugs ibuprofen and diclofenac on glioma (HTZ-349, A172 and U87MG) cell proliferation, migration and lactate formation. Similar to ibuprofen, diclofenac treatment resulted in migration decrease in a time- and concentration-dependent manner, better expressed in HTZ-349 and A172 cells. Diclofenac and ibuprofen induced cell cycle arrest in three cell lines, although at different checkpoints: at the G2/M check-point and in S-phase for diclofenac and at the G1/G0 state in all cell lines for Ibuprofen. Gottfried et al. [Citation16] showed that diclofenac targets MYC and glucose metabolism in tumour cells thus inhibiting their proliferation and migration. Similarly, Seliger et al. [Citation37] established a relation between diclofenac-reduced extracellular lactate levels and the significant reduction in glioma cell migration. We also observed decreased migration of diclofenac-treated tumour cells. The most significant reduction in tumour cell migration after treatment with diclofenac was established for HeLa cervical adenocarcinoma cells. The migration of HT-29 colorectal carcinoma and MCF-7 breast adenocarcinoma cells was also inhibited by diclofenac treatment, however, the effect was less pronounced.
There are multiple mechanisms of action proposed to explain the diverse anticancer effects of diclofenac [Citation7,Citation35,Citation36]. Our data confirm and complement the knowledge on the in vitro antitumor effects of diclofenac in three different permanent human cancer cell lines (HT-29; HeLa, and MCF-7). According to the results of this study, it can be supposed that diclofenac exerts its antitumor effect by inhibiting cell proliferation and migration, and induction of apoptotic death.
Conclusions
The present study showed that the three examined cancer cell lines (HeLa, HT-29 and MCF-7) were sensitive to diclofenac. The effect of diclofenac on the cell viability and proliferation was more pronounced on the 48 h and strongest in MCF-7, followed by HT-29 and HeLa tumour cells. Cytomorphological analysis indicated that the antitumour effect of diclofenac was achieved by inducing cell death through apoptosis. The wound healing assay clearly showed the ability of diclofenac to inhibit the migration of the tested cancer cells. Our results show that diclofenac has anti-proliferative and proapoptotic effects that are in agreement with published data showing that NSAIDs inhibit the proliferation and impair the growth of different cultured cancer and noncancer cells. The observed antineoplastic effect of diclofenac requires further investigation of its potential utility in cancer therapy.
Disclosure statement
The authors report no conflicts of interest.
Data availability statement
All data that support the findings reported in this study are available from the corresponding author upon reasonable request.
References
- IQVIA 2020. Market Segmentation, MAT; Apr 2020. Available from: www.iqvia.com.
- Brogden R, Heel R, Pakes G, et al. Diclofenac sodium: a review of its pharmacological properties and therapeutic use in rheumatic diseases and pain of varying origin. Drugs. 1980;20(1):24–48.
- Gan TJ. Diclofenac: an update on its mechanism of action and safety profile. Curr Med Res Opin. 2010;26(7):1715–1731.
- Alfaro RA, Davis DD. Diclofenac. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020.
- Ghanghas P, Jain S, Rana C, et al. Chemopreventive action of non-steroidal anti-inflammatory drugs on the inflammatory pathways in colon cancer. Biomed Pharmacother. 2016;78:239–247.
- Cha YI, RN. DuBois NSAIDs and cancer prevention: targets downstream of COX-2. Annu Rev Med. 2007;58:239–252.
- Pantziarka P, Sukhatme V, Bouche G, et al. Repurposing Drugs in Oncology (ReDO)-diclofenac as an anti-cancer agent. Ecancermedicalscience. 2016;10:610.
- Rao P, Knaus EE. Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): cyclooxygenase (COX) inhibition and beyond. J Pharm Pharm Sci. 2008;11(2):81s–110s.
- Gunaydin C, Bilge SS. Effects of nonsteroidal anti-inflammatory drugs at the molecular level. Eurasian J Med. 2018;50(2):116–121.
- Dermond O, Rüegg C. Inhibition of tumor angiogenesis by non-steroidal anti-inflammatory drugs: emerging mechanisms and therapeutic perspectives. Drug Resist Updat. 2001;4(5):314–321.
- Zappavigna S, Cossu AM, Grimaldi A, et al. Anti-inflammatory drugs as anticancer agents. IJMS. 2020;21(7):2605.
- Hashemi Goradel N, Najafi M, Salehi E, et al. Cyclooxygenase-2 in cancer: a review. J Cell Physiol. 2019;234(5):5683–5699.
- Wang X, Baek SJ, Eling T. COX inhibitors directly alter gene expression: role in cancer prevention?Cancer Metastasis Rev. 2011;30(3–4):641–657.
- Tangrea JA, Albert PS, Lanza E, et al. Non-steroidal anti-inflammatory drug use is associated with reduction in recurrence of advanced and non-advanced colorectal adenomas (United States). Cancer Causes Control. 2003;14(5):403–411.
- Díaz-Rodríguez L, García-Martínez O, Morales MA, et al. Effects of indomethacin, nimesulide, and diclofenac on human MG-63 osteosarcoma cell line. Biol Res Nurs. 2012;14(1):98–107.
- Gottfried E, Lang SA, Renner K, et al. New aspects of an old drug-diclofenac targets MYC and glucose metabolism in tumor cells. PLoS One. 2013;8(7):e66987.
- Hixson LJ, Alberts DS, Krutzsch M, et al. Antiproliferative effect of nonsteroidal antiinflammatory drugs against human colon cancer cells. Cancer Epidemiol Biomarkers Prev. 1994;3(5):433–438.
- Arisan ED, Ergül Z, Bozdağ G, et al. Diclofenac induced apoptosis via altering PI3K/Akt/MAPK signaling axis in HCT 116 more efficiently compared to SW480 colon cancer cells. Mol Biol Rep. 2018;45(6):2175–2184.
- Al-Nimer MS, Hameed HG, Mahmood MM. Antiproliferative effects of aspirin and diclofenac against the growth of cancer and fibroblast cells: in vitro comparative study. Saudi Pharm J. 2015;23(5):483–486.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63.
- Nikolova I, Marinov L, Georgieva A, et al. Metamizole (dipyrone) - cytotoxic and antiproliferative effects on HeLa, HT-29 and MCF-7 cancer cell lines. Biotechnol Biotechnol Equip. 2018;32(5):1327–1337.
- Sudsai T, Wattanapiromsakul C, Nakpheng T, et al. Evaluation of the wound healing property of Boesenbergia longiflora rhizomes. J Ethnopharmacol. 2013;150(1):223–231.
- Arisan ED, Akar RO, Rencuzogullari O, et al. The molecular targets of diclofenac differs from ibuprofen to induce apoptosis and epithelial mesenchymal transition due to alternation on oxidative stress management p53 independently in PC3 prostate cancer cells. Prostate Int. 2019;7(4):156–165.
- Yang L, Li J, Li Y, et al. Diclofenac impairs the proliferation and glucose metabolism of triple‑negative breast cancer cells by targeting the c‑Myc pathway. Exp Ther Med. 2021;21(1):1–9.
- Yagi K, Kawasaki Y, Nakamura H, et al. Differential combined effect of COX inhibitors on cell survival suppressed by sorafenib in the HepG2 cell line. Biol Pharm Bull. 2014;37(7):1234–1240.
- Leidgens V, Seliger C, Jachnik B, et al. Ibuprofen and diclofenac restrict migration and proliferation of human glioma cells by distinct molecular mechanisms. PLoS One. 2015;10(10):e0140613.
- Chirasani SR, Leukel P, Gottfried E, et al. Diclofenac inhibits lactate formation and efficiently counteracts local immune suppression in a murine glioma model. Int J Cancer. 2013;132(4):843–853.
- Zerbini LF, Tamura RE, Correa RG, et al. Combinatorial effect of non-steroidal anti-inflammatory drugs and NF-κB inhibitors in ovarian cancer therapy. PLoS One. 2011;6(9):e24285.
- Valle BL, D’Souza T, Becker KG, et al. Non-steroidal anti-inflammatory drugs decrease E2F1 expression and inhibit cell growth in ovarian cancer cells. PLoS One. 2013;8(4):e61836.
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516.
- Gomez-Lechon MJ, Ponsoda X, O’Connor E, et al. Diclofenac induces apoptosis in hepatocytes by alteration of mitochondrial function and generation of ROS. Biochem Pharmacol. 2003;66(11):2155–2167.
- Kusuhara H, Komatsu H, Sumichika H, et al. Reactive oxygen species are involved in the apoptosis induced by nonsteroidal anti-inflammatory drugs in cultured gastric cells. Eur J Pharmacol. 1999;383(3):331–337.
- Hickey EJ, Raje RR, Reid VE, et al. Diclofenac induced in vivo nephrotoxicity may involve oxidative stress-mediated massive genomic DNA fragmentation and apoptotic cell death. Free Radic Biol Med. 2001;31(2):139–152.
- García-Martínez JM, Fresno Vara JA, Lastres P, et al. Effect of metamizol on promyelocytic and terminally differentiated granulocytic cells: comparative analysis with acetylsalicylic acid and diclofenac. Biochem Pharmacol. 2003;65(2):209–217.
- Inoue A, Muranaka S, Fujita H, et al. Molecular mechanism of diclofenac-induced apoptosis of promyelocytic leukemia: dependency on reactive oxygen species, Akt, Bid, cytochrome and caspase pathway. Free Radic Biol Med. 2004;37(8):1290–1299.
- Albano F, Arcucci A, Granato G, et al. Markers of mitochondrial dysfunction during the diclofenac-induced apoptosis in melanoma cell lines. Biochimie. 2013;95(4):934–945.
- Seliger C, Leukel P, Moeckel S, et al. Lactate-modulated induction of THBS-1 activates transforming growth factor (TGF)-beta2 and migration of glioma cells in vitro. PLoS One. 2013;8(11):e78935.