
Abstract
Contemporary genetic methods have not yet solved the ‘missing heritability’ problem of complex diseases such as Alzheimer’s disease (AD). The impact of rare or less common variation on human complex diseases and traits remains to date barely investigated. In this study rare population variants detected using whole-exome sequencing were employed to examine how molecular pathways are prioritized in four groups: Alzheimer’s disease (AD) patients, Frontotemporal dementia (FTD) patients, young and healthy individuals and centenarians. The set of prioritized genes in AD patients associated with Semaphorin interactions pathways, contrasting with the results of the other groups. We identified rare pathogenic, likely pathogenic and variants of unknown significance in these prioritized genes in AD patients. The results of this study offer evidence that semaphorin pathways play a role in AD genetic etiology.
Introduction
Pathological changes in the brain structure may commence twenty years before the clinical manifestations of Alzheimer’s disease (AD) [Citation1–4]. At the onset of the clinical phase, which could last years, mild symptoms of cognitive impairment (MCI) develop as a result of synapse loss [Citation5]. Neuron loss continues as the disease progresses resulting in 15–40% loss in hippocampal volume and in inflammatory processes in hippocampus and cerebral cortex [Citation6]. These neurodegenerative processes have severe clinical manifestations, e.g. impaired memory, cognition and language, and in due course lead to dementia [Citation7, Citation8].
Some of the pathological alterations that occur in the brain are associated with proteopathies. According to the ‘amyloidogenic hypothesis’ the accumulation of amyloid plaques in the extracellular matrix triggers a cascade of pathological processes leading to neuronal dysfunction and cell death [Citation9]. Complex molecular interactions among different immune responses and disorders of regulatory pathways in synapses have been established as causes for AD [Citation10, Citation11].
Studies on AD patients have revealed a large number of pathogenic fully penetrant mutations in the genes APP, PSEN1, PSEN2 causing autosomic dominant forms of AD. The ε4 allele of the АРОE gene is a major genetic risk factor for both early- and late-onset AD [Citation12]. Mutations in MAPT and TREM2 genes have also been confirmed as risk factors [Citation13]. Massive parallel resequencing techniques have established at least 21 additional loci associated with AD [Citation14, Citation15].
The SORL1 gene encodes the Sortilin-related receptor LR11/SorLA involved in the control of amyloid beta peptide production. Studies have observed increased risk for developing late-onset and autosomal dominant early-onset AD associated with common and rare SORL1 variants [Citation16]. These variants are associated with AD as they likely disrupt the processing of the APP proteins [Citation17, Citation18]. A study on a Belgian cohort of AD patients found that several ABCA7 gene loss-of-function mutations were in higher frequency in the patients compared to a control group, and one frameshift mutation (Glu709fs) was found to segregate with disease in a family with autosomal dominant inheritance of Alzheimer’s disease [Citation19].
The methods that are widely used in the postgenomic era, i.e. case/control genome-wide association studies and whole genome/exome sequencing of families, have not solved the problem of ‘missing heritability’, i.e. a large number of variants predisposing to complex diseases remains undiscovered. It has been proposed for several polygenic traits that rare variants contribute to a substantial proportion of this ‘missing heritability’ [Citation20, Citation21]. The role of rare and low-frequency genetic variants (MAF < 1%) in complex diseases is however hard to establish, as they are usually left statistically undetected in association studies [Citation22–24].
The impact of rare or less common gene variants on complex human diseases and traits remains therefore to date unclear. Target sequencing performed on Chinese AD families found a number of rare coding variants in risk genes for neurodegenerative diseases to be significantly associated with AD, including rare variants in PLD3, LRRK2, ABCA7, TREM2 and FUS genes [Citation25]. A search for low-frequency coding variants with large effects on late-onset AD in risk families identified a rare variant in the PLD3 gene which increased the risk for late-onset AD twofold, along with evidence that the PLD3 gene influences APP processing [Citation26]. Genome-wide searches have established rare variants in relatively few genes, e.g. TREM2, AKAP9, UNC5C, ZNF655, IGHG3 and CASP7, to be associated with AD [Citation27].
Studying individual genes however cannot reveal the complex interactions among them. It is not yet clear how rare coding variants alter the protein structure and function and trigger the pathogenic mechanisms of neurodegenerative disorders. We speculate that the effect of common variants is combined with that of rare variants in molecular pathways and alter biological processes. Determining the role of Mendelian and non-Mendelian diseases in molecular pathogenesis is also hindered by lack of functional or family history data. In silico predictions, well-established American College of Medical Genetics and Genomics (ACMG) criteria for variant classification, and allele frequency considerations are tools that are used to determine variant pathogenicity [Citation28]. The ACMG criteria include: population frequencies of variants in genome databases, computational and in silico predictions, functional data, segregation analysis in family pedigrees, allelic data and patient’s phenotype.
The aim of the present study was to explore the role of rare variants, obtained by whole-exome sequencing (WES), in biological processes and molecular pathways leading to AD pathogenesis. Our analysis is based on comparing the WES results of AD patients with those of Frontotemporal dementia (FTD) patients, a centenarian group, and young and healthy controls.
Subjects and methods
Ethics statement
This study was approved by the Ethics committee of the Medical University of Sofia, adhering to national and international legislation.
Subjects
AD and FTD patients had been diagnosed by a team of clinical neurologists using standard diagnostic approaches. Compiling a control group for this study was complicated by the fact that the age of AD onset varies widely, and that it is difficult to predict the risk for healthy individuals to develop the disease at a later age. We choose therefore to use two types of controls, i.e. young and healthy individuals and Bulgarian centenarians. Individuals with no AD symptoms were sequenced in the centenarian group, after they were examined by a clinical team that included gerontologists in order to assess their physical and mental status. A questionnaire was devised to get information about their lifestyle, medical history, neurological status, movement independence, cardiovascular disease, cancer, diabetes, etc. The control group was constituted of young healthy individuals, free of acute or chronic diseases.
DNA extraction and sequencing
Whole-exome sequencing was performed on four DNA pools composed with equimolar amounts of DNA extracted from: 66 AD patients, 70 FTD patients, 32 Bulgarian centenarians (100–106 years old) and 61 young and healthy individuals (25–30 years old). DNA was extracted using QIAamp DNA Blood Mini Kit (Qiagen). AD and FTD patients’ pools were whole exome sequenced using Agilent SureSelect Human All ExonV6 kit (Agilent Technologies, CA, USA), whereas the centenarian and young control pools were sequenced using BGI v4 chemistry on a BGISEQ-500 platform (BGI Genomics) at a mean 250× coverage. Such high coverage is required for pool-seq sequencing to ensure that alleles with low frequency are also detected [Citation29].
Bioinformatics analysis
Following the ‘best practice’ recommendations for pool-seq data, we initially filtered all pools as follows: total depth of coverage ≥ 30, mapping quality ≥ 60, supporting base quality ≥ 30, number of reads per MAF ≥ 2, fraction of observations supporting an alternate allele ≥ 0.0001. For further analyses we selected exonic and splicing variants, frameshift/non-frameshift insertions, non-synonymous SNVs, startloss, stopgain and stoploss variants. As we based our analysis on variants with very low population frequencies, we selected variants with European population frequencies MAF ≤ 0.001 (non-Finnish Europeans genome [Citation30]), but enhanced in the AD and FTD pools (MAF ≥ 0.01). From the centenarian and young control pools were selected variants with MAF ≤ 0.01 and number of reads per MAF ≥ 4. The Varsome database was used to classify variants according to their clinical significance [Citation31]. The REACTOME online platform [Citation32] was used to establish and to graphically visualize molecular pathways that might be affected by the set of genes carrying rare variants.
Results
The whole-exome sequencing established 303,135–368,090 variants in the four pools analyzed. The selected genes with rare population variants were 2299 in the AD patients’ pool, 2221 in the FTD patients’ pool, 2685 in the centenarian pool and 2605 in the control group of young and healthy individuals. Prioritized pathways related to developmental biology for Alzheimer’s patients contrasted to the other analyzed clinical and control groups ():
Figure 1. REACTOME pathways associated with Development Biology in: AD patients (А); FTD patients (В); Centenarians (С) and Young controls (D). The numbers denote enhanced pathways: 1. Semaphorin interactions, 2. NCAM1 interaction, 3.Interaction between La and Ankyrins, 4. Formation of cornified envelope, 5. Keratinization.
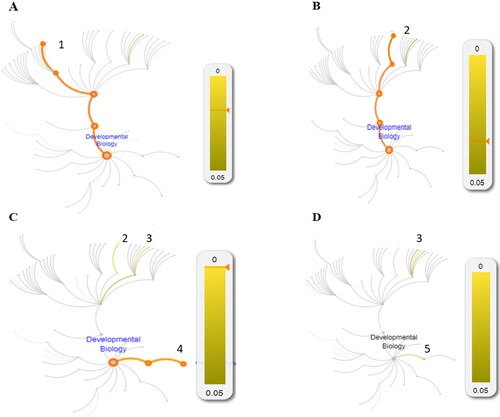
In AD patients the Semaphorin interactions pathway is prioritized (p-value 1.8E-2; FDR 9.65E-1);
In the FTD patients NCAM1 interactions pathway is prioritized (p-value 3.63E-2; FDR 9.47E-1);
In the centenarian group NCAM1 interactions (p-value 1.64E-2; FDR 9.13E-1), Interaction between L1 and Ankyrins (p-value 1.37E-2; FDR 9.13E-1) and Formation of the cornified envelope pathways are prioritized (p-value 6.11E-4; FDR 6.17E-1),
In the young controls’ group Interaction between L1 and Ankyrins pathway is prioritised (p-value 2.51E-2; 9.46E-1) and Keratinization (p-value 2.68E-2; FDR 9.46E-1).
The prioritization of the Semaphorin interactions pathway in AD patients is due to enrichment of 11 genes – PLXND1, PLXNA1, PLXNA4, PLXNB3, PLXNC1, HOMER3, SEMA5A, SEMA5B, SEMA6A, PTPRC and TREM2.
The gene TREM2 contains the pathogenic variant rs104894002 estimated to be with 0.016 frequency in AD patients, whereas its population frequency is 9 × 10−6. This gene also contains two rare variants classified as variants of unknown significance (VUS): rs201258314 and rs374851046. The PLXND1 gene contains the missense variant rs746887877 enriched in AD patients and six VUSs (rs748102549, rs762249042, rs867360787, rs762376973, rs373770425 and rs750005799). In five additional genes are detected VUSs: PLXNA1 (rs777776780), PLXNA4 (rs186378769), PLXNB3 (rs781784671), PLXNC1 (rs189549726) and HOMER3 (rs141596306). The variants in the remaining four genes (PLXNA1, SEMA5A, SEMA5B and SEMA6A) are presumably benign.
The seven genes with pathogenic and VUSs were input in the REACTOME platform in an effort to obtain a more accurate picture of the molecular pathways associated with Semaphorin interactions pathway in AD patients ( and ).
Figure 2. Molecular pathways indicated by the seven genes with pathogenic and VUSs.
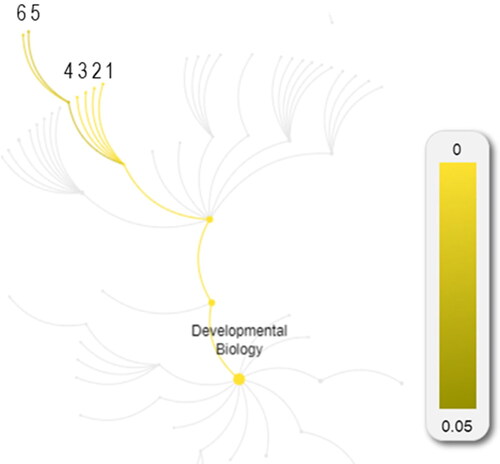
Table 1. Significant pathways associated with developmental biology in AD patients.
Discussion
The aim of the present study was to examine if and how rare variants impact gene prioritization of molecular pathways in AD patients, FTD patients, centenarians with no AD symptoms and young and healthy individuals. Our results suggest that in AD patients enriched genes associate with Semaphorin interactions, molecular pathways constituent of the nervous system development events.
Changes in neural connectivity and loss of synaptic contacts are an indication of neurodegenerative disease pathogenesis. It has been suggested that aberrant semaphorin expression or function may result in altered neuronal connectivity or synaptic function associated with a number of developmental and degenerative neural disorders [Citation33]. Such role for semaphorins is most strongly supported by experimental data for AD and ALS (amyotrophic lateral sclerosis). Compelling evidence for semaphorins playing a role in AD is also provided by the discovery of a multiprotein complex from brain tissue of AD patients that contains the phosphorylated microtubule-associated proteins 1B, Sema3A, CRMP-2, plexinA1 and -A2 [Citation34]. Semas and their related receptors have been implicated in developmental and adult onset nervous system diseases including AD [Citation35].
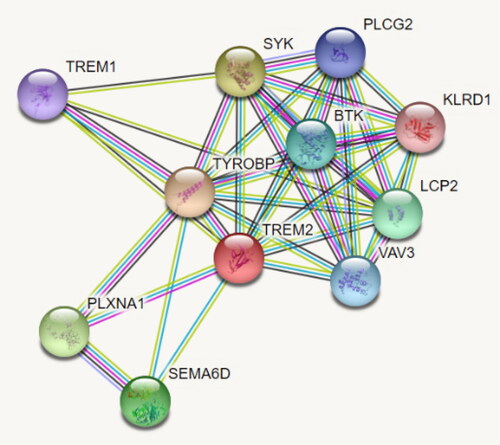
Growing axons are highly adaptable at the growing point, which senses environmental guidance cues and responds by undergoing cytoskeletal changes. Semaphorins are highly conserved families of guidance molecules that guide axons [Citation36]. Semaphorins signal via receptor complexes that include other proteins such as plexins. Interactions between different subfamilies of plexins and semaphorins show differential specificity and trigger different sets of biological functions [Citation37]. Our results show that AD patients carry the pathogenic variant rs104894002 in the gene TREM2, previously established in another study of AD patients [Citation38], along with two rare VUSs: rs201258314 and rs374851046. TREM2 gene mutations have been shown to increase the risk of neurodegenerative conditions such as AD, ALS and Parkinson’s disease [Citation27]. The TREM2 protein is a part of a complex network, including PLXNA1 and the semaphorin SEMA6D (). Additional studies are needed in order to elucidate which rare pathogenic variants and rare VUSs variants play a role in AD genetic etiology. It is conceivable that such variants in the TREM2 gene, in interaction with PLXNA1 and SEMA6D variants, are implicated in molecular pathways associated with AD pathogenesis.
Figure 3. Molecular network involving the TREM2 protein.
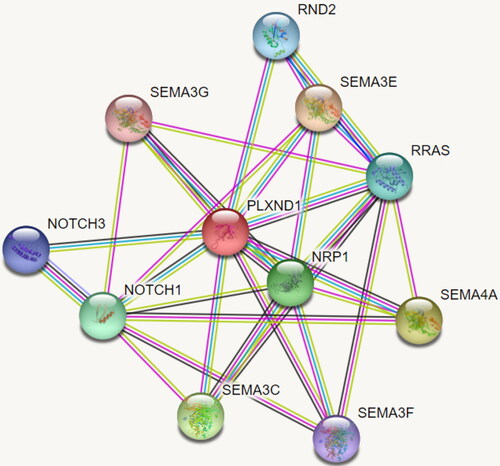
The PLXND1 gene accommodates the likely pathogenic variant rs746887877 along with 6 VUSs. This gene plays an important role in cell–cell signaling, in regulating the migration of many cell types and in ensuring the specificity of synapse formation. Plexin-D1 is a cell surface receptor for the proteins SEMA4A, SEMA3C, SEMA3F, SEMA3E and SEMA3G (). Enriched rare VUSs are found in semaphore genes associated with PLXND1, e.g. SEMA4A, SEMA3C, SEMA3F and SEMA3G. It can be speculated that likely pathogenic variants and an array of VUSs in the PLXND1 gene, along with the VUSs in the semaphoring genes, have the potential to disrupt crucial biological processes in the brain leading to AD.
Figure 4. The molecular network which the PLXND1 gene is part of.
It is worth mentioning that microRNAs (miRNAs) play a key role in growth, differentiation and cell death by suppressing one or more target genes. The activity of miRNAs in the brain is affected by various factors such as electromagnetic fields, diminishing the significance of AD associated variants that might be carried the subject. There is evidence that long-term exposure to certain radiofrequencies alters miRNA expression in the brain and may lead to adverse effects such as development of neurodegenerative diseases, e.g. AD, but the evidence for this is inconclusive and more studies should be devoted on this topic [Citation39, Citation40].
Conclusions
Our results suggest differential molecular pathway prioritization in AD patients compared to FTD patients, centenarians with no AD symptoms and young and healthy individuals. The presence of rare pathogenic, likely pathogenic and variants of unknown significance in genes associated with semaphorin interactions in AD patients suggest the involvement of these pathways in the complex picture of genetic disturbances in the molecular pathogenesis of Alzheimer’s disease.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work is funded by the National Science Fund of Bulgaria, project KP-06-N33/5 from 13.12.2019.
Data availability statement
Data are available upon reasonable request from the authors.
References
- Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357–367.
- Reiman EM, Quiroz YT, Fleisher AS, et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol. 2012;11(12):1048–1056.
- Jack CR, Jr, Lowe VJ, Weigand SD, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: implications for sequence of pathological events in Alzheimer’s disease. Brain. 2009;132(5):1355–1365.
- Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367(9):795–804.
- Mufson EJ, Binder L, Counts SE, et al. Mild cognitive impairment: pathology and mechanisms. Acta Neuropathol. 2012;123(1):13–30.
- Vijayakumar A, Vijayakumar A. Comparison of hippocampal volume in dementia subtypes. ISRN Radiol. 2013;2013:174524.
- Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21(3):383–421.
- Wyss-Coray T, Lin C, von Euw D, et al. Alzheimer’s disease-like cerebrovascular pathology in transforming growth factor-beta 1 transgenic mice and functional metabolic correlates. Ann N Y Acad Sci. 2000;903:317–323.
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595–608.
- Criscuolo C, Fabiani C, Cerri E, et al. Synaptic dysfunction in Alzheimer’s disease and glaucoma: from common degenerative mechanisms toward neuroprotection. Front Cell Neurosci. 2017;11:53.
- Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30(4):572–580.
- Liu C-C, Liu C-C, Kanekiyo T, et al. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9(2):106–118.
- Benitez BA, Cooper B, Pastor P, et al. TREM2 is associated with the risk of Alzheimer’s disease in Spanish population. Neurobiol Aging. 2013;34(6):1711.e15–1711.e17.
- Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of alzheimer disease: clinical implications and perspectives. Genet Med. 2016;18(5):421–430.
- Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. 2015;77(1):43–51.
- Verheijen J, Van den Bossche T, van der Zee J, et al. A comprehensive study of the genetic impact of rare variants in SORL1 in European early-onset Alzheimer’s disease. Acta Neuropathol. 2016;132(2):213–224.
- Vardarajan BN, Zhang Y, Lee JH, et al. Coding mutations in SORL1 and Alzheimer disease. Ann Neurol. 2015;77(2):215–227.
- Pottier C, Hannequin D, Coutant S, et al. High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol Psychiatry. 2012;17(9):875–879.
- Cuyvers E, De Roeck A, Van den Bossche T, et al. Mutations in ABCA7 in a Belgian cohort of Alzheimer’s disease patients: a targeted resequencing study. Lancet Neurol. 2015;14(8):814–822.
- Gutierrez-Rodrigues F, Calado RT. The interpretation of rare or novel variants: damaging vs. disease-causing. Rev Bras Hematol Hemoter. 2018;40(1):3–4.
- Bomba L, Walter K, Soranzo N. The impact of rare and low-frequency genetic variants in common disease. Genome Biol. 2017;18(1):77.
- Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753.
- Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11(6):415–425.
- Jordan B. [Common and rare variants, polygenic traits and missing heritability]. Med Sci. 2017;33(6-7):674–676.
- Zhang W, Jiao B, Xiao T, et al. Association of rare variants in neurodegenerative genes with familial Alzheimer’s disease. Ann Clin Transl Neurol. 2020;7(10):1985–1995.
- Cruchaga C, Karch CM, Jin SC, et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature. 2014;505(7484):550–554.
- Patel D, Mez J, Vardarajan BN, et al. Association of rare coding mutations with Alzheimer disease and other dementias among adults of European ancestry. JAMA Netw Open. 2019;2(3):e191350.
- Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19(1):4–23.
- Fracassetti M, Griffin PC, Willi Y. Validation of pooled whole-genome re-sequencing in Arabidopsis lyrata. PLoS One. 2015;10(10):e0140462.
- Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443.
- Kopanos C, Tsiolkas V, Kouris A, et al. VarSome: the human genomic variant search engine. Bioinformatics. 2019;35(11):1978–1980.
- Jassal B, Matthews L, Viteri G, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020;48(D1):D498–D503.
- Pasterkamp RJ, Giger RJ. Semaphorin function in neural plasticity and disease. Curr Opin Neurobiol. 2009;19(3):263–274.
- Good PF, Alapat D, Hsu A, et al. A role for semaphorin 3A signaling in the degeneration of hippocampal neurons during Alzheimer’s disease. J Neurochem. 2004;91(3):716–736.
- Uchida Y, Ohshima T, Sasaki Y, et al. Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3β phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes Cells. 2005;10(2):165–179.
- Stoeckli ET. Understanding axon guidance: are we nearly there yet?Development. 2018;145(10):dev151415.
- Gay CM, Zygmunt T, Torres-Vázquez J. Diverse functions for the semaphorin receptor PlexinD1 in development and disease. Dev Biol. 2011;349(1):1–19.
- Lu Y, Liu W, Wang X. TREM2 variants and risk of Alzheimer’s disease: a meta-analysis. Neurol Sci. 2015;36(10):1881–1888.
- Dasdag S, Akdag MZ, Erdal ME, et al. Effects of 2.4 GHz radiofrequency radiation emitted from Wi-Fi equipment on microRNA expression in brain tissue. Int J Radiat Biol. 2015;91(7):555–561.
- Dasdag O, Adalier N, Dasdag S. Electromagnetic radiation and Alzheimer’s disease. Biotechnol Biotechnol Equip. 2020;34(1):1087–1094.