
Abstract
Epigenetics plays an important role in plant resistance to biotic and abiotic stresses, although few studies have focused on soybean epigenetics associated with cadmium stress. In this study, hydroponically grown soybean seedlings were treated with hydroponic nutrient solution containing various concentrations of CdCl2 (0.5, 5, 15 mg L−1) to investigate the effects of cadmium exposure on soybean genomic DNA methylation levels and patterns using methylation-sensitive amplified polymorphism (MSAP) analysis. Overall, the results revealed alterations of genomic DNA methylation levels and patterns in soybean seedlings under cadmium stress, with methylation level and methylation polymorphism rate both increasing in a dose-dependent manner with increasing cadmium concentration. The total methylation rates for genomic DNA of control (CK), S1 (0.5 mg L−T CdCl2), S2 (5 mg L−1 CdCl2) and S3 (15 mg L−1 CdCl2) groups were 27.92%, 29.78%, 33.2% and 35.79%, respectively, and methylation polymorphism rates were 14.46%, 16.57% and 18.15%, respectively. Thus, soybean cadmium stress-induced methylation changes mainly reflected increased genomic DNA methylation. After polymorphic methylated genomic DNA fragments were recovered from polyacrylamide gels, cloned and sequenced, sequence homology comparisons revealed that among 30 detected differentially methylated fragments, 15 sequences were highly homologous to genes encoding functional proteins already known to be associated with plant stress responses. These results indicate that soybean resists cadmium stress by increasing both the level of genomic DNA methylation and the genomic DNA remethylation rate. This work provides a methodological foundation for investigating molecular mechanisms underlying the soybean response to cadmium stress.
Introduction
Soybeans (Glycine max) are an important source of high-quality protein and vegetable oil, which both greatly benefit global human health and survival. However, abiotic stresses such as drought, high salinity, low temperature and exposure to heavy metals often lead to reduced soybean yields and crop losses [Citation1]. Cadmium, a particularly harmful heavy metal and the most common heavy metal pollutant in farmland soil, has known detrimental effects on soybean growth, crop yield and quality. After cadmium is absorbed by plants, it accumulates in roots, stems, leaves and other tissues. Plants grown in soil with high cadmium levels exhibit root growth inhibition, plant dwarfism and chlorosis that directly compromise crop yield and quality [Citation2].
DNA methylation, an important mechanism used by eukaryotic cells to regulate gene expression, maintain genome stability and sustain normal growth and development, has received considerable attention in recent years. In plants and many other eukaryotes, expression of genes within heterochromatin domains is determined in part by methylation of cytosines at position 5 of the pyrimidine ring (5-Me-C), with methylation of 20%–30% of cytosine residues in plant genomes typically observed under normal growth conditions [Citation3]. Once DNA methylation of a given genomic cytosine residue occurs, it can be sustained for many generations as an epigenetic change that can trigger stress-induced gene evolution [Citation4, Citation5]. Thus, stress induced by external environmental changes can lead to genomic DNA methylation site alterations that can alter the expression patterns of functionally adaptive genes. Therefore, the study of genomic DNA methylation changes that occur during the soybean response to cadmium exposure would enhance our understanding of key genes and molecular pathways involved in soybean adaptation to cadmium stress. This knowledge would be useful for guiding development of new soybean varieties with greater resistance to cadmium stress.
Methylation-sensitive amplified polymorphism (MSAP) analysis, based on amplified fragment length polymorphism (AFLP) analysis, is a common method used for generating and analyzing whole-genome methylation profiles. It has advantages of simple operation, high polymorphism-detection rate and effectiveness in detecting large numbers of cytosine methylation changes occurring at CCGG sites throughout an entire genome. In plants, this method has been effectively used to investigate methylation changes occurring in genomes of corn, rice, sorghum, pepper and other crops [Citation6–9]. Specifically, MSAP technology has been used for detecting increases in genomic methylation level, upregulation of methyltransferase gene expression and nuclear chromatin structural changes in aquatic plants under cadmium stress. Such results have demonstrated that some plants can adapt to cadmium stress by regulating chromatin structure. In turn, chromatin structure can be controlled by altering genomic DNA methylation patterns within specific chromatin regions to control the balance between gene expression and gene silencing for groups of genes [Citation10]. In this work, these methods were used to evaluate changes in genomic methylation profiles of soybean seedlings after exposure to cadmium stress, since few investigations with this focus have been reported to date.
In this study, in order to reveal epigenetics-based molecular mechanisms underlying the soybean response to cadmium stress, MSAP technology was used to analyze overall genomic DNA methylation levels and modes of methylation. Moreover, functional annotation analysis of DNA sequences corresponding to differentially methylated genomic DNA fragments obtained from plants under cadmium stress revealed the identities and putative functions of numerous genes with altered expression associated with cadmium stress. These results provide a methodological foundation to guide future studies of soybean cadmium stress response mechanisms. This information will be useful for guiding the development cadmium-resistant soybean varieties.
Materials and methods
Plant materials, enzymes and reagents
The soybean variety ‘Heinong 48’ was provided by Heilongjiang Academy of Agricultural Sciences. For 115 days from emergence to maturity, the active accumulated temperature should be gricultural Scienc2380 °C. It is an early maturing and high protein soybean variety. It has long leaves, purple flowers, gray hairs and light brown pods. The plant is 80–95 cm high, with 17 nodes of main stem and branches. The seeds are round, yellow and glossy, and the hilum is yellow. The 100 seeds weight is 22–25 g. From late April to early May, the yield per hectare is about 2480 480. From late April to early May, the yield per .
Table 1. Main reagents used in this study.
Treatments and experimental grouping
Fully formed soybean seeds of uniform size were selected, soaked in distilled water for 30 min then soaked in 0.1% NaCl solution for 30 min. Next, seeds were sown in sterilized vermiculite in plastic pots that were placed in a greenhouse under controlled light and temperature (25 °C) conditions. After cotyledons fully emerged from seeds (with flattened appearance), plants were irrigated with 0.5× Hoagland nutrient solution every 2 days then were allowed to grow until the third compound leaf pair was visible. Next, soybean seedlings were selected based on similarity of growth vigor then selected seedlings were transferred to glass jars containing 1× Hoagland nutrient solution for cultivation.
Next, seedlings in jars were randomly assigned to the various treatment groups, with 3 jars used for each treatment (4 plants per jar). The nutrient solution was changed daily during treatment. After 24 h of culture in 1× Hoagland nutrient solution, seedlings were treated with or without CdCl2 at various concentrations then plants were harvested after 8 days of treatment. Each treatment was as follows: Control group (CK): 1× Hoagland nutrient solution + sprayed distilled water; S1: 1× Hoagland nutrient solution + sprayed 0.5 mg L−1 CdCl2; S2: 1× Hoagland nutrient solution + sprayed 5 mg L−1 CdCl2; S3: 1× Hoagland nutrient solution + sprayed 15 mg L−1 CdCl2.
Extraction of soybean genomic DNA
After 8 days of growth, soybean seedlings were rinsed with deionized water then 0.1 g of soybean leaves was cut from the same node of each plant in each group (CK, S1, S2 and S3) then each leaf fragment was placed into a mg L-1 centrifuge tube and frozen in liquid nitrogen. DNA was extracted using the SDS method [Citation11, Citation12], then for each sample the DNA concentration was determined and the purity was assessed using a UV spectrophotometer and 1% agarose gel electrophoresis, respectively.
Methylation-sensitive amplified polymorphism (MSAP) analysis
The methylation-sensitive amplified polymorphism (MSAP) analysis procedure used here was based on the method reported by Pan Yajiao et al. [Citation13], with modifications. Methylation-sensitive restriction enzymes HpaII and MspI were combined with restriction enzyme EcoRI (E + H, E + M) then genomic DNA for each experimental group was double-digested with each pair of enzymes. Sequences of adaptors and pre-amplification and selective amplification primers are shown in . The reaction system and reaction conditions are shown in . After a DNA denaturing step, PCR products were separated by size using 2% polyacrylamide gel electrophoresis, with detection of bands conducted using 0.1% silver nitrate staining and images recorded via digital imaging and electrophoretic bands recording.
Table 2. Adapter and primer sequences.
Table 3. Reaction system in the analysis of methylation sensitive amplified polymorphism (MSAP).
Data collection
After conducting polyacrylamide gel electrophoresis and staining to reveal different reaction product bands, numbers of electrophoretic bands within each lane for each primer combination were counted. The presence of a band was recorded as a ‘1’ and absence of a band was recorded as a ‘0’ and results of triplicate experiments were analyzed to determine the statistical significance.
Analysis of DNA methylation level and pattern change
Sensitivities of homolyase HpaII and MspI to methylation status of DNA restriction sites differ such that MSAP analysis of double-digested restriction fragments can reveal the methylation status of an individual CCGG site, as well as the overall degree of restriction site methylation throughout an entire genome. DNA samples were digested with EcoRI/HpaII(H) and EcoRI/MspI (M), resulting in generation of four types of methylation band patterns (Types I-IV). For a Type I band pattern, bands were found in both H and M gel lanes, indicating that there was no methylation at the CCGG-site. For a type II band pattern, a band was found in the H-lane but not in the M-lane, indicating that single strand lateral methylation (hemimethylation/CHG methylation) occurred at the CCGG-site. For a type III band pattern, no band was found in the H-lane, but a band was found in the M-lane, indicating that the CCGG-site had undergone double-stranded medial methylation (full methylation/CG methylation). For a type IV band pattern, no band was found in the H-lane or M-lane, indicating that the CCGG-site had undergone double-stranded lateral methylation. Ultimately, only 3 types band patterns (type I, II and III) were detected on polyacrylamide gels in this study.
Results and discussion
Effects of cadmium stress on DNA methylation levels
For whole-genome MSAP analysis, 42 primer pair combinations were used for analyzing the soybean DNA methylation patterns in CK (control) S1 (0.5 mg L−1), S2 (5 mg L−,) and S3 (15 mg L−1) samples, with relative frequencies of amplified bands analyzed statistically between groups. It can be seen from that under Cd stress, MSAP absent target ratios (that indicate highly methylated sites) of soybean DNA of seedlings treated with S1, S2 and S3 (29.78%, 33.2%, 35.79%, respectively) were higher than the corresponding CK group ratio (27.92%). Total methylation (double-chain methylation) rates were 22.37%, 23.9%, 25.2% and 25.73% and half methylation rates were 5.55%, 5.89%, 8% and 10.06%, respectively. Thus, Cd stress exposure of groups S1, S2 and S3 appeared to lead to increased genomic DNA cytosine methylation, causing the rate of full methylation to exceed the half methylation rate. Thus, from these results it can be inferred that the predominant methylation status of CCGG sites in the genomes of the soybean plants under Cd stress was that of full methylation (double-stranded). Moreover, as compared with the control group, the total methylation level of soybean increased after Cd exposure such that as Cd concentration increased, the total DNA methylation level in soybean leaves gradually increased, indicating the existence of a significant dose-dependent relationship between the increase in total methylation level and Cd concentration.
Table 4. Effects of cadmium stress on soybean leaf genomic DNA methylation level.
Plants are known to adapt to external environmental stresses by employing mechanisms that involve epigenetic modifications (such as DNA methylation). DNA methylation is thought to be a relatively conserved form of epigenetic modification that plays important roles in maintaining normal plant growth and development and in plant adaptation to adverse external environmental conditions, although a change in methylation level usually has a negative impact on plants [Citation14]. It has been suggested that the transcriptional initiation interaction between promoter and RNA polymerase is interrupted if the cytosine in the promoter region of the coding gene is methylated, resulting in gene expression silencing, as evidenced by observations that demethylation of gene promoters can activate genes and promote gene expression [Citation15]. Thus, investigating changes in plant genome DNA methylation levels and patterns would greatly enhance our understanding of molecular and metabolic mechanisms underlying plant stress resistance, growth and development. In turn, such knowledge could be used to guide the development of soybean varieties with greater resistance to cadmium stress.
When plants are under heavy metal stress, they can regulate gene expression by altering the methylation of genomic DNA sites so as to silence or activate specific genes in order to cope with the stress [Citation16]. In fact, studies have demonstrated that heavy metal stress is directly related to altered plant genomic DNA methylation levels and methylation patterns [Citation17]. For example, after exposures to cadmium and lead stresses, the total methylation levels of rice and wheat genomes increased [Citation17, Citation18], while the genomic DNA methylation levels in rapeseed seedlings after cadmium and chromium stress exposures exceeded the corresponding control levels and were positively correlated with the metal concentrations [Citation19]. Conversely, during heavy metal stress due to exposures to heavy metals Ni, Cd and Cr, the changes in the overall cytosine methylation levels of other plant species, such as clover and hemp, decreased [Citation20]. Here our results showed that the total methylated genomic DNA content of soybean seedlings after exposure to different Cd concentrations increased on average as compared with the control, with the total methylation level increasing in a dose-dependent manner with increasing Cd concentration. More specifically, although the total methylation level of soybean genomic DNA treated with 0.5 mg L− Cd (29.78%) was barely greater than the control level (27.92%), the total genomic DNA methylation levels of soybean plants treated with 5 mg L−C and 15 mg L−a Cd were significantly greater than the control levels, as observed in rapeseed [Citation19]. It has been suggested that plants may maintain genome stability through methylation to avoid oxidative damage caused by heavy metal stress [Citation21]. In this study, the rates of total methylation and semi methylation of soybean genomic DNA in the control group were 22.37% and 5.55%, respectively, while the corresponding rates for plants exposed to Cd at a concentration of 0.5 mg L−. were 23.9% and 27.73%, respectively, and to Cd at a concentration of 15 mg L−2 were 5.89% and 10.06%, respectively. These results thus indicated that altered methylation of soybean genomic DNA in response to plant exposure to Cd was mainly due to full methylation of CCGG sites. Nevertheless, the change in the semi methylation level after Cd exposure exceeded that of full methylation, indicating that under Cd stress the soybean genome tended to undergo unstable hemimethylation as an important protective mechanism to cope with stress.
Effects of cadmium stress on DNA methylation patterns
By comparing the MSAP results for the soybean plants under Cd stress and the results for the control group, 15 band pattern differences were detected ( and , ). It can be seen from and that the numbers of type A (methylation type) loci in the genomic DNA of S1, S2 and S3 Cd-exposed plant groups were 182, 232 and 273, respectively, accounting for 7.74%, 9.9% and 11.69% of the total methylated polymorphic loci, respectively. Type B (demethylation type) loci numbers of 150, 149 and 143 were detected, respectively, accounting for 6.38%, 6.35% and 6.12% of total methylated polymorphic loci, respectively. Type C (indefinite type) loci numbers accounted for 0.34% of the total methylated polymorphic loci. Type D (with unchanged methylation status) polymorphic rate accounted for 85.54%, 83.43% and 81.85% of the total methylation polymorphic loci, respectively. As compared with CK (control), the methylation polymorphism rates for S1, S2 and S3 Cd-treated groups were 14.46%, 16.57% and 18.15%, respectively. It can be seen that with increasing Cd concentration, the methylation polymorphism rates of soybean genomic DNA increased, while the proportion of demethylated sites remained almost unchanged. Meanwhile, the number of methylated polymorphic sites gradually increased and ultimately exceeded the numbers of demethylated polymorphic sites. Therefore, the ratio of methylation to demethylation (net methylation) of soybean genomic DNA increased with increasing Cd concentration, indicating that high-level Cd exposure led to an increase in total methylation level of soybean genomic DNA.
Figure 1. Soybean leaf DNA methylation banding patterns. H1–H4: EcoR I+Hpa II combination; M1–M4: EcoR I+Msp I combination; 1: Control group leaf; 2: S1 group leaf; 3: S3 group leaf; 4: S4 group leaf; D: Monomorphism, D1: no methylation occurred, D2: Hemimethylation, D3: total methylation; A: Methylation, A1, A2 and A3: Remethylation, A4 and A5:Hypermethylation; B: Demethylation, B1, B2, B3, B4 and B5: Demethylation ; C: Uncertain type, C1 and C2: Uncertain type.
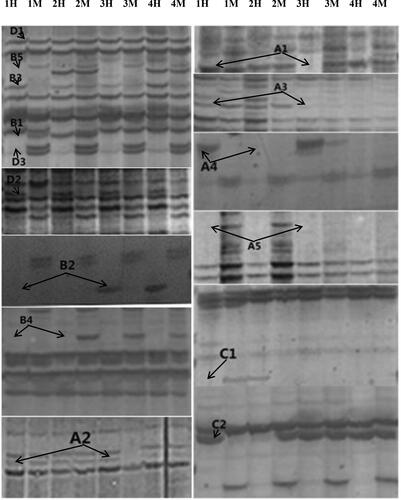
Table 5. Changes of methylation pattern of soybean genome under cadmium stress.
Table 6. Effects of cadmium stress on soybean leaf genomic DNA methylation.
Plant regulation of gene expression mainly employs two methylation modes, remethylation and demethylation, with a change in the methylation mode in vivo involved in the protection of plants from stress. In this study, MSAP was conducted using 42 pairs of primers to amplify soybean genomic DNA, with results showing increasing soybean genomic DNA methylation polymorphism rates with increasing Cd concentration (0.5, 5 and 15 mg L−i), with methylation ratios of 7.74%, 9.9% and 11.69% and demethylation ratios of 6.38%, 6.35% and 6.12% observed, respectively. Thus, these results demonstrated that the main DNA methylation mode change in the soybean genome after Cd exposure mainly involved DNA remethylation.
In higher plants, DNA methylation pattern changes vary according to the type of heavy metal stress; for example, specific genomic DNA sequences in root cells of clover and marijuana exposed to heavy metals Ni, CD or Cr tend to undergo demethylation [Citation20]. Conversely, methylation of some plant gene promoters and coding region sequences can silence the expression of associated genes during normal plant growth and development [Citation22]. Usually, demethylation of plant DNA is thought to activate the expression of genes that induce synthesis of protective enzymes to resist harmful environmental conditions, since methylation cannot activate gene expression but can inhibit the transcription and expression of some genes. Nevertheless, after plants are exposed to certain stressors, their genomes can undergo remethylation as a defensive mechanism, mainly because methylation can inhibit the transcription of transposons and exogenous DNA and thus reduce the risk of damage resulting from genomic non-allelic translocation and recombination [Citation23]. For example, Luo Jun et al. [Citation24] showed that when different varieties of maize were subjected to Pb stress, DNA methylation patterns of resistant maize materials tended to be biased toward remethylation, while maize materials with lower resistance tended to be biased toward demethylation. As another example, Yang Jinlan et al. [Citation25] found that under Cd stress, radish plants mainly initiated a dynamic Cd stress response that was biased toward demethylation to counter toxic Cd effects. In this study, absent target ratios of DNA methylation polymorphism after soybean seedling exposures to 0.5, 5, and 15 mg L−1 Cd were 14.46%, 16.57% and 18.15%, respectively. Thus, the DNA methylation mode changes were mainly due to DNA methylation that may have shut down the expression and inhibited the transcription of some related genes through methylation, thus enhancing the resistance of soybean plants to toxic Cd exposures.
Sequencing and homology analysis of methylated DNA fragments
To identify the functions of the proteins encoded by differentially methylated genomic fragments sequenced in this work, the NCBI Blast search tool (http://www.ncbi.nlm.nih.gov/) was used for conducting Blast homology searches of DNA sequences obtained from 30 differentially methylated bands. The results () showed that 9 of 30 sequences encoded proteins that were homologous with proteins in the database with unknown functions (E3, E7, E11, E12, E16, E18, E22, E24, E28). Meanwhile, six fragments encoded proteins with no homologues in the database (E6, E13, E15, E21, E23, e25), while the remaining 15 sequences had high homology with genes encoding proteins with known functions as follows: CCR3 serine/threonine-protein kinase (E1), wnk8 serine/threonine-protein kinase (E26), ZF HD homologous protein (E4), envelope protein (E8), atscl13 protein (member of GRAS family) (E27), malic acid transporter (E29), glycosamine-rich RNA binding protein (E2), antisense mediated mRNA degradation regulator (Upf1) (E5), GDP-l-fucosynthase (E10), protein translation initiation factor Sui1 (E14, E30), yipf1 (E19, E20) homologous protein, galactosaldehyde (E9) and microtubulin (E17). Notably, analysis of the functions of these proteins indicated they were mainly associated with cell metabolic pathways and plant stress resistance-associated pathways.
Table 7. Blast result of DNA differential methylation bands.
Functional cluster analysis revealed that proteins encoded by 15 differentially methylated genomic DNA fragments had homology to proteins with known functions that were associated with plant responses to salt, drought, low temperature and heavy metal stresses. For example, the UPF1 protein is an important regulator of nonsense-mediated mRNA decay (NMD). NMD selectively degrades mRNA that contains premature translation termination codons (PTCs) to minimize cell damage caused by accumulation of C-terminally truncated proteins and is a key mechanism for controlling post-transcriptional mRNA quality in eukaryotic cells [Citation26]. PTCs, which mainly result from DNA- or RNA-level base changes that introduce translational stop codons into mRNA, are detected by UPF1 factor, the substrate of NMD [Citation27]. A combination of alternative splicing (AS) and nonsense-mediated RNA degradation (AS/NMD) is employed as a post-transcriptional monitoring mechanism for regulating gene expression. Studies have shown that AS/NMD can induce the expression and transcription of many genes, including transcription factors and stress response genes that participate in normal plant growth, development and adaptation to stresses, [Citation28], with mRNA degradation playing an essential role in maintaining plant health and vigor. In this work we found that demethylation of a gene homologous to UPF1 (E5) may play a role in the transcriptional regulation of mRNA degradation factor induced by plant exposure to cadmium as an adaptive mechanism employed by soybean plants to resist cadmium stress [Citation29].
Conclusions
The total methylation and methylation polymorphism ratios of soybean genomic DNA increased after cadmium exposure in a dose-dependent manner with increasing Cd concentration triggering a change in methylation mode that increased genomic DNA methylation overall. Of the 30 differentially methylated DNA fragments characterized here, 15 had sequences that were highly homologous to genes encoding proteins with known functions that were mainly associated with plant stress responses. Strategies targeting such genes may be useful for developing soybean varieties with greater resistance to cadmium stress.
Ethics statement
There are no researches conducted on animals or humans.
Acknowledgements
We hereby thank the Heilongjiang Academy of Agricultural Sciences for providing the soybean material for breeding.
Data availability statement
The data that support the findings of this study are available from the authors upon reasonable request.
Disclosure statement
There are no conflicts to declare.
Funding
This study was supported by the Harbin Science and Technology Innovation Talent Research Project (RC2013QN002103), the Heilongjiang Provincial Postdoctoral Funding Project (LBH-Z13185), the Heilongjiang Provincial Department of Education Science and Technology Research Project (12511154) and the Harbin Normal University Doctoral Research Fund (KGB200903), Harbin Normal University graduate innovative scientific research project “Cloning and functional study of soybean squalene coxidase GmSQE3 gene”, Postgraduate research project of Harbin Normal University (project number: HSDSSCX2021-03; grant ID 10,000RMB).
Funding
The author(s) reported there is no funding associated with the work featured in this article.
References
- Fan JP, Bai X, Li Y, Ji W, Wang X, Cai H, Zhu YM. Cloning and functional analysis of wild soybean S-adenosylmethionine synthase gene. Acta Agron Sin. 2008;(9):1581–1587. (In Chinese with English abstract).
- Wang H, Cuiping H, Nan Z. Effects of cadmium on growth characteristics and biomass of lawn plants. Pratacult Sci. 2003; 20(5):32–34. (In Chinese with English abstract).
- Bender J. DNA methylation and epigenetics. Annu Rev Plant Biol. 2004; 55:41–68.
- Ganesan A. Epigenetics: the first 25 centuries. Philos Trans R Soc London Ser B Biol Sci. 2018; 373(1748).
- Soodeh T, Jacqueline B. DNA methylation: toward crop disease resistance improvement. Trends Plant Sci. 2019;24(12):1137–1150.
- Tan MP. Analysis of DNA methylation of maize in response to osmotic and salt stress based on methylation-sensitive amplified polymorphism. Plant Physiol Biochem. 2010;48(1):21–26.
- Xiong LZ, Xu CG, Maroof MAS, et al. Patterns of cytosine methylation in an elite rice hybrid and its parental lines, detected by a methylation-sensitive amplification polymorphism technique. Mol Gen Genet. 1999;261(3):439–446.
- Zhang MS, Xu CM, Yan HY, et al . Limited tissue culture-induced mutations and linked epigenetic modifications in F hybrids of sorghum pure lines are accompanied by increased transcription of DNA methyltransferases and 5-methylcytosine glycosylases. Plant J. 2009; 57(4):666–679.
- Portis E, Acquadro A, Comino C, et al. Analysis of DNA methylation during germination of pepper (capsicum annuum L) seeds using methylation-sensitive amplification polymorphism(MSAP). Plant Sci. 2004;166(1):169–178.
- Greco M, Chiappetta A, Bruno L, et al. In Posidonia oceanica cadmium induces changes in DNA methylation and chromatin patterning. J Exp Bot. 2012; 63(2):695–709.
- Wang X, Teng D, Tian F, et al. Comparison of three DNA extraction methods for feed products and four amplification methods for the 5’-junction fragment of roundup ready soybean. J Agric Food Chem. 2012;60(18):4586–4595.
- Jingjing M. Study on the mechanism of gene silencing by affecting epigenetic modification in arabidopsis mat4 [D]. China Agricultural University, 2018. (In Chinese with English abstract).
- Pang YJ, Fu BY, Wang D, et al. Analysis of the temporal and spatial changes of DNA methylation induced by rice drought stress. Sci Agric Sin. 2009;42(09):3009–3018. (In Chinese with English abstract).
- Zhang YL, Ye WW, Wang JJ, et al. Research progress of DNA methylation and plant stress resistance. Northwest Botany. 2009; 29(07):1479–1489. (In Chinese with English abstract).
- Vanyushin BF, Ashapkin VV . DNA methylation in higher plants: past, present and future. Biochim Biophys Acta. 2011;1809(8):360–368.
- Yu P, Gao F, Liu J, et al. Effects of cadmium on plant growth and research progress on the mechanism of plant cadmium tolerance. Chin Agric Sci Bull. 2017; 33(11):89–95. (In Chinese with English abstract).
- Huang ZX, Wang FJ, Jiang H, Ji ZL, Ding Y F, Jiang Q, Tao Y L, Zhu C. Expression of cadmium accumulation-related genes in two rice varieties and their molecular regulatory mechanisms. Acta Agron Sin. 2014;40(4):581–590. (in Chinese with English abstract).
- Ge CL, Yang XY, Liu XN, et al. Effect of heavy metal on levels of methylation in DNA of rice and wheat. Plant Physiol Mol Biol. 2002; 28:363–368. (In Chinese with English abstract).
- Labra M, Grassi F, Imazio S, et al . Genetic and DNA-methylation changes induced by potassium dichromate in Brassica napus L. Chemosphere. 2004;54(8):1049–1058.
- Aina R, Sgorbati S, Santagostino A, et al. Specific hypomethylation of DNA is induced by heavy metals in white clover and industrial hemp. Physiol Plant. 2004;121(3):472–480.
- Li XL, Lin ZX, Nie YC, et al. MSAP analysis of epigenetic changes in cotton genomic DNA under salt stress. Acta Agron Sin. 2009;35(4):588–596. (In Chinese with English abstract).
- Wassenegger M. RNA directed DNA methylation. Plant Mol Biol. 2000;43(2–3):203–220.
- Peng H, Xi T, Zhang J. Stability of plant DNA methylation under stress. Sci Agric Sin. 2011;44(12):2431–2438. (In Chinese with English abstract).
- Luo J. Study on Pb stress methylation model of maize inbred lines and determination of antioxidant enzyme activity. Sichuan Agricultural University; 2011. p. 1–53. (In Chinese with English abstract).
- Yang JL, Liu LW, Gong YQ, et al. Analysis of DNA methylation sensitive amplified polymorphism of radish genome under cadmium stress. Zhi Wu Sheng Li Yu Fen Zi Sheng Wu Xue Xue Bao. 2007; 33(3):219–226. (In Chinese with English abstract).
- Alper C, Feng H, Allan J. NMD monitors translational fidelity 24/7. Curr Genet. 2017;63(6):1007–1010.
- Jia XB, Hu J. Nonsense-mediated mRNA degradation. Chin J Biochem Mol Biol. 2012;28(2):115–120. (In Chinese with English abstract).
- Maria K, Craig GS, Naeem HS. Alternative splicing and nonsense-mediated decay modulate expression of important regulatory genes in arabidopsis. Nucleic Acids Res. 2012;40:2454–2469.
- Liang WX. The role of mRNA degradation in plant development and resistance response. Biotechnol Bull. 2012;(4):1–6. (In Chinese with English abstract).