
Abstract
NAD(P)H oxidases (NOXs) catalyze the oxidation of NAD(P)H by reducing molecular O2 to H2O2 or H2O. Water-forming NOXs have been highlighted as promising biocatalyst systems for regeneration of NAD+ or NADP+ in industrial applications. The NADH oxidase of Giardia lamblia (GlNOX) uses naturally both nicotinamide cofactors producing only water as byproduct; therefore, it can be considered a multipurpose and innocuous regeneration system for redox reactions using either NAD+ or NADP+. From a biotechnological perspective, kinetic stability (and not thermodynamic stability) must be the most important aspect to consider for enzymes involved in industrial applications; therefore, understanding the kinetic stability of a technologically interesting enzyme is an indispensable step toward its application. In this work, we analyze the thermal inactivation kinetics of GlNOX and the influence of diverse additives on it. A combination of 1,4-Dithiothreitol as reducing agent and the disaccharide trehalose protects remarkably GlNOX from inactivation. Structural studies indicate that inactivation is not related to denaturation, whereas overoxidation of the active site cysteine seems to be the main factor related to loss of the enzyme activity. It is concluded that understanding the factors that influence the protein inactivation of GlNOX allowed to improve its kinetic stability, making it biotechnologically valuable.
Introduction
Oxidoreductases are the largest group of enzymes in the Enzyme Commission Nomenclature and have broad applications in biotechnological process such as the oxidation of alcohols and amines, the stereoselective reduction of aldehydes and ketones and the production of chiral alcohols and amines by deracemization [Citation1]. The vast majority of known oxidoreductases use nicotinamide adenine dinucleotide (NAD+ or NADH) or nicotinamide adenine dinucleotide phosphate (NADP+ or NADPH) in its oxidized or reduced form depending on the electron transfer direction. For industrial applications, the use of equimolar concentrations of these cofactors to drive redox reactions to completion would be prohibitive owing to their costs. Therefore, it has been highlighted that efficient and economic regeneration systems are critical to make these processes economically viable [Citation2]. Chemical, electrochemical, photochemical and enzymatic methods for cofactor regeneration have been described [Citation3], but enzymatic methods are the most promising because of their high stereoselectivity and turnover numbers. In this connection, a NADH oxidase from the gram-positive bacterium Lactobacillus brevis was first suggested as a plausible biocatalyst for NAD+ regeneration [Citation4].
NAD(P)H oxidases (NOXs) are FAD-dependent oxidoreductases belonging to the flavoprotein disulfide reductase enzyme family [Citation5]; these catalyze the oxidation of NAD(P)H by reducing molecular oxygen by two-electron transfer producing hydrogen peroxide, or four-electron transfer forming water [Citation1]. NOXs can selectively use NADH or NADPH, but some enzymes can use both [Citation2]. As water-producing NOXs employ only oxygen as substrate and produce water as unique byproduct and not reactive oxygen species or its precursors, these enzymes are considered superior prospects for cofactor regeneration in biotechnological applications, mainly in the chemical industry [Citation6, Citation7]. For example, the utility of NOXs as regeneration systems has been demonstrated in the oxidation of 2-heptanol to 2-heptanone [Citation1], or in the conversion of glycerol to 1,3-dihydroxyacetone [Citation7].
We have previously characterized the recombinant NADH oxidase from the protozoan Giardia lamblia (GlNOX) [Citation8]. G. lamblia causes giardiasis, one of the most common parasitoses in humans. In addition to its medical significance, Giardia has many interesting biological characteristics; just to mention a few, it lacks peroxisomes, Golgi apparatus, typical mitochondria performing oxidative phosphorylation and common enzymes aimed to cope with oxidative stress (superoxide dismutase, catalase, peroxidase, glutathione peroxidase and glutathione reductase) [Citation8]. Given these features, G. lamblia is a fermentative-microaerophilic organism that relies on GlNOX activity to remove oxygen from the intracellular milieu in order to maintain the redox state and to regenerate oxidized nucleotides (NAD+ and NADP+) [Citation8]. Recombinant GlNOX was heterologously expressed in Escherichia coli and purified in a single step by immobilized metal affinity chromatography (IMAC) yielding a homogenous, pure and stable protein. The kinetic characterization of the enzyme shows that, unlike most NOXs, GlNOX can use both nicotinamide cofactors, albeit with preference for NADH over NADPH. The Km of GlNOX for NADH was ∼5-fold lower than that for NADPH (17.7 vs. 87.4 µmol/L, respectively), whereas the Vmax of the enzyme was almost double with NADH as compared to NADPH (63.4 vs. 36.4 µmol/(min mg)). GlNOX produces H2O but not H2O2; thus, it is catalogued within the group of water-forming NOXs. As GlNOX uses naturally both nicotinamide cofactors producing only water, it can be considered a multipurpose and innocuous regeneration system for redox reactions using either NAD+ or NADP+ [Citation8].
Protein stability is a major concern for biotechnological purposes since this determines the likelihood of an enzyme being able to be taken toward industrial applications; the greater the stability, the greater the possibility of being applied. The stability is related to its capacity to retain its biological function under diverse conditions, including non-physiological environments. Protein stability can be analyzed from two perspectives: thermodynamic stability or kinetic stability. Thermodynamic stability involves the energy necessary to go from the folded to the unfolded state under equilibrium conditions. Kinetic stability on the other hand is related to the time-dependent irreversible inactivation in going from a full-active state to partially or totally inactive states. Therefore, kinetic stability (preservation of the active state) and not thermodynamic stability (protein unfolding) is considered the most important process to consider for enzymes involved in industrial applications [Citation9–11].
In this work we analyze the thermal inactivation kinetics of GlNOX and the influence of diverse additives on it. The results shed light on the enzyme’s inactivation mechanism and how to improve its kinetic stability, making it biotechnologically valuable.
Materials and methods
Buffers, salts, substrates and reagents were purchased from Sigma-Aldrich. Recombinant GlNOx was produced as previously described [Citation8]. Briefly, the GlNOX gene cloned in the pET23b vector was expressed in E. coli BL21(DE3)pLysS cells and purified by IMAC. The activity of the enzyme was measured spectrophotometrically at 25 °C by following the oxidation of NADH as reported [Citation8], except that the reaction medium was saturated with medical-grade oxygen instead of air-saturated. Reaction media (1 mL) contained 0.2 NADH and 2 mmol/L DTT in 50 mmol/L Tris–HCl pH 8.0, 50 mmol/L NaCl [Citation8]. Protein concentration was determined by the Bradford method as described [Citation8].
Inactivation assays were performed by incubating the enzyme (1 mg/mL) at 50 °C in Tris-HCl 50 mmol/L, NaCl 50 mmol/L, pH 7.5 and following the residual activity in a temporal course. Experiments were performed in the absence or in the presence of the following additives: FAD 1 mmol/L, NAD+ 2 mmol/L, 1,4-Dithiothreitol (DTT) 20 mmol/L, trehalose 500 mmol/L and trehalose 500 mmol/L plus DTT 20 mmol/L. For specific ligands the concentrations used were suprastoichiometric (∼1:50 and ∼1:100 for FAD and NAD+, respectively); whereas DTT and trehalose were added at concentrations usually employed in this kind of assays [Citation12]. The linear sequential model of Henley and Sadana for enzyme inactivation was used to analyze the data [Citation13]. The general model involves the native enzyme (E) and active alternate forms of the enzyme (E1, E2 … En), which are in equilibrium. The general mathematical equation for this model is described by (AR = A1 e-k1t + A2 e-k2t + … An e-knt). For GlNOX all the inactivation curves were biphasic and therefore fitted to a double exponential decay model (AR = A1 e-k1t + A2 e-k2t) where AR is the residual activity, A1 and k1 are the apparent amplitude and rate constant for the first phase, respectively, whereas A2 and k2 are the apparent amplitude and rate constant for the second phase. A1, A2, k1 and k2 contain information about the real rate constants of the system; the population of the full-active enzyme at time t depends on the equilibria between the full-active, partially active or inactive states and therefore on the rate constants associated with its interconversion [Citation12, Citation13]. The reversibility of thermal inactivation was assayed by incubating GlNOX at the times necessary to reach approximately 75, 50 and 25% of residual activity; thereafter, samples were incubated at 4 °C in an ice-water bath and the activity followed as stated for 30 min.
Structural studies were performed as follows: enzyme samples (protein 1 mg/mL, Tris-HCl 50 mmol/L NaCl 50 mmol/L pH 7.5) without any treatment, heated for 30 min at 50 °C or incubated with 4 mol/L of urea for 2 h were analyzed by circular dichroism (CD) and fluorescence spectroscopy. Far-UV CD spectra were obtained from 190 to 280 nm (except for the urea spectra, which were stopped at 205 nm by the increase in voltage and noise signal beyond this point) in a Jasco J-810 spectropolarimeter. Assays were performed in a quartz cell with a path length of 0.1 cm at a protein concentration of 0.1 mg/mL. Intrinsic protein fluorescence spectra were obtained from 300 to 450 nm in a Perkin-Elmer LS-55 fluorescence spectrometer after excitation at 280 nm. Assays were conducted in a quartz cell with a path length of 1 cm with a protein concentration of 0.1 mg/mL. FAD fluorescence spectra were obtained from 480 to 620 nm after excitation at 450 nm in a quartz cell with a path length of 1 cm and a protein concentration of 0.1 mg/mL. All spectroscopic assays were performed at 25 °C. In all conditions, blanks were subtracted from the experimental samples.
Data analysis
All experiments were performed by at least three independent assays. For kinetic assays, each point represents the mean ± standard deviation (bars) of three independent measurements; data were fitted to the double exponential decay model described above by using the damped least-squares method. For spectroscopic assays, each spectrum was the average of three complete scans.
Results
The inactivation kinetics of GlNOX showed a biphasic pattern (, panel A), with a fast phase twenty-times faster than the slow phase (0.53 vs 0.026 min−1, respectively), and a slight preponderance of the first phase (57%) with the respect to the second one (44%) (). In order to start to define a possible model for GlNOX inactivation, the reversibility of the inactivation across the reaction progress was tested. Regardless of the grade of inactivation, the process was irreversible ().
Figure 1. (A) Inactivation kinetics of GlNOX (1 mg/mL) at 50 °C in Tris-HCl 50 mmol/L, NaCl 50 mmol/L pH 7.5. Buffer was preincubated for 10 min in an Eppendorf Thermomixer set at this temperature without mixing; thereafter, the enzyme was added and aliquots were withdrawn from the sample at the times indicated in the abscissa axis to measure the residual activity. Each point represents the mean ± standard deviation (bars) of three independent measurements. Initial activity at time 0 was considered 100%; relative activities at times n were calculated as the fraction of the initial activity and expressed as percentage. The line represents the fit of the data to a double exponential decay model as indicated in the Material and Methods section. (B) Inactivation kinetics of GlNOX in the same conditions as in panel A; except that FAD 1 mmol/L was added. (C) Inactivation kinetics of GlNOX in the same conditions as in panel A; except that NAD+ 2 mmol/L was added. (D) Inactivation kinetics of GlNOX in the same conditions that panel A; except that DTT 20 mmol/L was added. (E) Inactivation kinetics of GlNOX in the same conditions as in panel A; except that trehalose 500 mmol/L was added. (F) Inactivation kinetics of GlNOX in the same conditions that in panel A; except that trehalose 500 mmol/L and DTT 20 mmol/L were added.
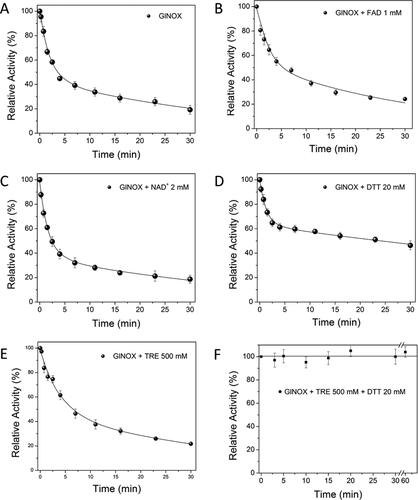
Figure 2. Reversibility of the thermal inactivation kinetics of GlNOX; the experiment was performed in the same conditions as in ; individual samples were incubated to reach approximately 75, 50 and 25% of residual activity, then these were transferred to an ice-water bath and the activity of each sample followed for 30 min.
Table 1. Kinetic constants for the thermal inactivation of GlNOX in the absence and in the presence of additives.
We hypothesize that by supplementing the inactivation reaction media with different additives, the inactivation kinetics of GlNOX could be modified, thus giving insights into the factors involved in the inactivation process. Since the FAD cofactor is not covalently bound to GlNOX, it was possible that the release of the coenzyme from the protein could be related to the inactivation process. To test this hypothesis, the same initial experiment of thermal inactivation of GlNOX was repeated, except that FAD 1 mmol/L was added to the incubation sample (, panel B). The inactivation kinetics of GlNOX in this condition was only slightly modified. The amplitude of the phases was reversed (44 vs 54% for the first and second phase, respectively) but the rate constants remained without significant changes ().
As ligand binding can change the stability of a protein, the same thermal inactivation experiment was performed in the presence of NAD+, the reaction product of the GlNOX reaction (, panel C). The ligand increases slightly the amplitude and the velocity of the first phase, whereas the amplitude of the second phase decreases but without changes in its velocity constant (). By the same token, it is well established that overoxidation of the active-site cysteine can inactivate many members of the NOX family [Citation1]; thus, the effect of DTT on the inactivation kinetics of GlNOX was tested. DTT changes the inactivation pattern of the enzyme significantly (, panel D); interestingly, the velocity of the first phase increases appreciably, but the velocity of the second phase decreases drastically. Furthermore, the amplitude of the fast phase decreases, whereas that of the slow phase increases considerably ().
As thermal protein denaturation could induce enzyme inactivation, the effect of trehalose on the inactivation kinetics of GlNOX was tested. Trehalose (α-D-glucopyranosyl-(1→1)-α-D-glucopyranoside) is an extensively described protein structure stabilizer [Citation14], with broad applications in biotechnological processes [Citation15]. The addition of trehalose to the GlNOX sample slowed down the inactivation of GlNOX, but to a lesser extent than expected (, panel E): the phases showed minimal changes and the velocity of the slow phase remains the same; only the velocity of the first phase decreases by half (). The most interesting result obtained arose by using the most active agents (DTT and trehalose) in combination (, panel F). As can be observed, the inactivation process completely stopped in this condition and the enzyme activity remained the same even if the incubation time was doubled. As activity decay was not observed, the kinetic constants were not calculated, but the effect of changing the medium composition on the kinetic behavior is quite apparent.
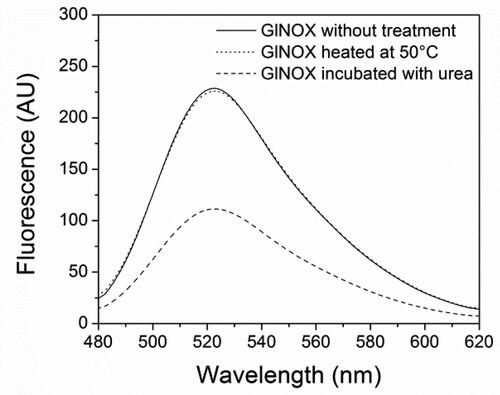
In order to examine deeply the inactivation process of GlNOX, structural studies were performed. Far-UV CD spectroscopy () shows that after 30 min at 50 °C, (where the enzyme has lost 80% of its activity), minor structural changes have occurred in the protein architecture, as can be inferred by the small changes in the CD spectra of the heated protein. Conversely, upon mild denaturation with urea, the changes in the CD spectra are appreciable and no enzyme activity could be detected. In the same way, protein intrinsic fluorescence shows similar results; the spectra of control and heated samples are similar, whereas in the urea treated sample the change in the fluorescence is considerable, both in the fluorescence intensity as in the maximum wavelength (). A very interesting result arose from the study of the protein-bounded FAD fluorescence. In the control and heated samples, the FAD spectra are almost superimposable, whereas in the sample incubated in the presence of urea, the intensity fluorescence of FAD decreases by half ().
Figure 3. Circular dichroism spectra of GlNOX without treatment, heated at 50 °C for 30 min or incubated with urea 4 mol/L for 2 h.
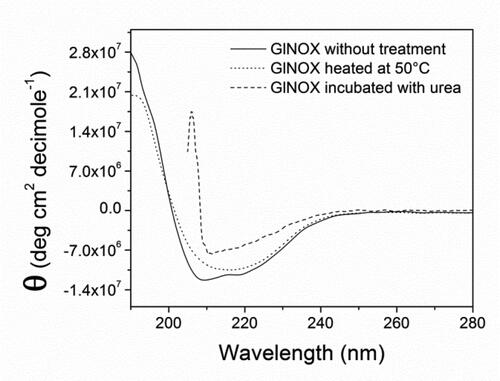
Figure 4. Intrinsic protein fluorescence of GlNOX without treatment, heated at 50 °C for 30 min or incubated with urea 4 mol/L for 2 h.
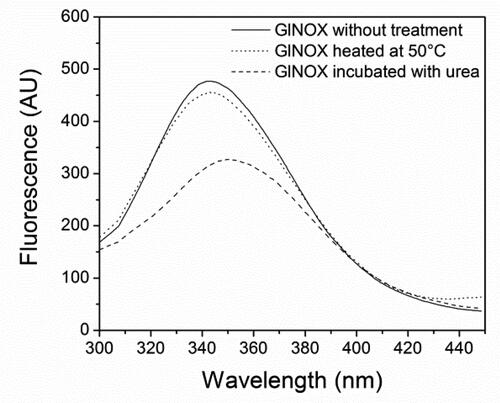
Figure 5. Fluorescence spectra of FAD bound to GlNOX without treatment, heated at 50 °C for 30 min or incubated with urea 4 mol/L for 2 h.
Discussion
According to the experimental evidence shown in this work ( and ), the thermal inactivation of GlNOX involves a two-state non-reversible model. Therefore, the inactivation of GlNOX under the experimental conditions tested can be schematized as N→N*→I, where N is the full active native enzyme, N* a partially active intermediate state and I the inactivated enzyme; k1 is the rate constant from N to N* and k2 the rate constant from N* to I; the reverse steps (k-1 and k-2), can be considered negligible. Supplementing the inactivation media with additives changes the inactivation kinetic constants of GlNOX, providing information on the main factors related to thermal inactivation, as stated below.
The minimal effect of FAD addition on the inactivation kinetics of GlNOX indicates that FAD release from the enzyme is not a relevant event at least under the experimental conditions tested in this work (, panel B and ). It has been reported that boiling of GlNOX for ten minutes is necessary to release the cofactor from this protein [Citation8, Citation16], implicating that FAD is strongly bound to GlNOX and destruction of the protein structure is necessary to disengage the cofactor. In contrast, the water-forming NOX-1 and NOX-2 from Bacillus cereus lost FAD during its purification, indicating that the cofactor is loosely bound to these enzymes [Citation17]. Along this line, it has been reported that the addition of exogenous FAD improves the activity of purified NOXs from Lactococcus lactis [Citation18] and Streptococcus faecalis [Citation19], which strongly suggests that the FAD cofactor is loosely bound to them too. Altogether, these results implicate that for GlNOX, the release of FAD from the enzyme is not related to inactivation.
It is generally accepted that ligand binding to a protein can change its stability; a ligand binding preferentially to a native protein will increase its stability, conversely, a ligand that binds preferentially to non-native states of the protein will decrease its stability [Citation20]. The effect of NAD+ on the global kinetic stability of the enzyme was negligible (, panel C and ); the addition of NAD+ to the enzyme shows marginally negative effects on the enzyme, slightly accelerating the inactivation process. It can be inferred that occupancy of the enzyme by the active-site ligand has minor effects on the kinetic stability of the enzyme.
The importance of cysteine oxidation on the kinetic stability of GlNOX was reinforced by the effect of DTT on the inactivation kinetics of the enzyme; as intuited, the reducing agent decreases significantly the inactivation of GlNOX (, panel D and ). It is proposed that the catalytic mechanism of the NOX family involves a redox-active cysteine residue (Cys42), which alternates between the thiol/thiolate and the sulfenic acid states during turnover [Citation21]. This cysteine residue can be overoxidized to sulfinic (–SO2H) and then to sulfonic acid (–SO3H) leading to inactivation [Citation22]. A group of NADH oxidases is dependent on the presence of a reducing agent for its optimal performance [Citation16, Citation22, Citation23], while others NOXs do not [Citation24–26]. The experimental data strongly suggest that the addition of DTT to the incubation medium reduces the overoxidation of the catalytic cysteine on GlNOX, thus slowing down its inactivation.
Protein denaturation seems to be an obvious factor related to enzyme thermal inactivation; therefore, the effect of trehalose, a known protein-structure stabilizer, was tested on GlNOX (, panel E and ). The effect of trehalose on the inactivation kinetics of GlNOX was less than expected, as changes as large as 30-times on the lifetime for thermal enzyme inactivation have been reported in response to the addition of trehalose [Citation12]. The result indicates that protein denaturation seems to play a minor role on the inactivation kinetics of GlNOX.
The additive effect of the most active agents used in combination (DTT and trehalose, , panel F) indicates that these act synergistically. The individual effect of DTT is not unexpected, and for trehalose it was less than expected. It is presumed that the small effect of trehalose on enzyme inactivation (, panel E and ) can be related to a local rearrangement that makes the active site cysteine less prone to oxidation, but that in the presence of DTT protects GlNOX completely from inactivation (, panel F). This assumption is supported by the structural data shown here. These data indicate that at conditions where the catalytic activity has been reduced by ∼80% (, panel A), the secondary and tertiary structure of the protein has been barely affected, as minimal changes in the circular dichroism and the intrinsic fluorescence of the protein are observed ( and ). In the same way, the fluorescence of the bound cofactor shows no changes in this condition, indicating that the structure of the active site is conserved (). On the contrary, under mild chemical denaturing conditions with urea, structural changes are clearly observed, as shown by the changes in the circular dichroism, intrinsic fluorescence spectra and in the FAD fluorescence, which indicate loss of secondary and tertiary structures and changes in the active site of the enzyme ().
It has been previously shown that for some enzymes, inactivation takes place before major conformational changes occur [Citation27], as shown here for GlNOX. As indicated above, some members of the NOX family require a reducing milieu to avoid oxidative inactivation [Citation16, Citation22, Citation23], whereas others do not [Citation24–26]. This implicates that even in the case of the same catalytic mechanism, the local conditions around the catalytic cysteine can change its propensity to overoxidation. In this way, it is feasible that a small conformational change around the catalytic site on GlNOX avoids the oxidation of the Cys42 residue in the presence of DTT, which in turn results in the increased kinetic stability shown here. The result is interesting because trehalose has been pointed out as a strong stabilizer of biomolecules both in vivo and in vitro, whose properties have been extensively reviewed [Citation15, Citation28] and with wide applications spanning from the pharmaceutical, food and cosmetic industries to cell, tissue and organs preservation [Citation15]. In addition, trehalose has been approved by the European Medicines Agency and the Food and Drug Administration as a safe ingredient [Citation29]. Finally, it has been shown that structure stabilizing molecules as trehalose can be used as reaction additives in order to stabilize enzyme activity at high temperatures, thus increasing its potential for biological, medical and industrial applications [Citation30]. In fact, in polymerase chain reaction (PCR) reactions, DTT and trehalose enhanced the amplification efficiency by stabilizing Taq DNA polymerase and lowering the DNA melting temperature [Citation31].
As GlNOX is an enzyme able to use naturally both cofactors (NADH and NADPH) with no side products other than water and is naturally more stable that other similar water-forming NOXs, those from Lactobacillus pentosus for example [Citation7], GlNOX seems to be a better prospect for biotechnological purposes.
Conclusions
GlNOX can be used as a safe and universal regeneration system for redox reactions. The main factor related to the kinetic stability of GlNOX is overoxidation of the active site cysteine, rather than protein denaturation. The kinetic stability of GlNOX can be improved by additives to increase its biotechnological potential.
Acknowledgments
The authors thank Sara-Teresa Méndez (Laboratorio de Bioquímica-Genética, Instituto Nacional de Pediatría) for technical support.
Conflicts of interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Data availability statement
The data that support the findings of this study are available from the corresponding author, [JOH], upon reasonable request.
Additional information
Funding
References
- Petschacher B, Staunig N, Müller M, et al. Cofactor specificity engineering of Streptococcus mutans NADH oxidase 2 for NAD(P)(+) regeneration in biocatalytic oxidations. Comput Struct Biotechnol J. 2014;9:e201402005.
- Gao H, Li J, Sivakumar D, et al. NADH oxidase from Lactobacillus reuteri: a versatile enzyme for oxidized cofactor regeneration. Int J Biol Macromol. 2019;123:629–636.
- Zhang YW, Tiwari MK, Gao H, et al. Cloning and characterization of a thermostable H2O-forming NADH oxidase from Lactobacillus rhamnosus. Enzyme Microb Technol. 2012;50(4–5):255–262.
- Geueke B, Riebel B, Hummel W. NADH oxidase from Lactobacillus brevis: a new catalyst for the regeneration of NAD. Enzyme Microb Technol. 2003;32(2):205–211.
- Argyrou A, Blanchard JS. Flavoprotein disulfide reductases: advances in chemistry and function. Prog Nucleic Acid Res Mol Biol. 2004;78:89–142.
- Li FL, Zhou Q, Wei W, et al. Switching the substrate specificity from NADH to NADPH by a single mutation of NADH oxidase from Lactobacillus rhamnosus. Int J Biol Macromol. 2019;135:328–336.
- Zhang JD, Cui ZM, Fan XJ, et al. Cloning and characterization of two distinct water-forming NADH oxidases from Lactobacillus pentosus for the regeneration of NAD. Bioprocess Biosyst Eng. 2016;39(4):603–611.
- Castillo-Villanueva A, Méndez ST, Torres-Arroyo A, et al. Cloning, expression and characterization of recombinant, NADH oxidase from Giardia lamblia. Protein J. 2016;35(1):24–33.
- Xie Y, An J, Yang G, et al. Enhanced enzyme kinetic stability by increasing rigidity within the active site. J Biol Chem. 2014;289(11):7994–8006.
- Iyer PV, Ananthanarayan L. Enzyme stability and stabilization – aqueous and non-aqueous environment. Process Biochem. 2008;43(10):1019–1032.
- Eijsink VG, Bjørk A, Gåseidnes S, et al. Rational engineering of enzyme stability. J Biotechnol. 2004;113(1–3):105–120.
- Guerrero-Mendiola C, Oria-Hernández J, Ramírez-Silva L. Kinetics of the thermal inactivation and aggregate formation of rabbit muscle pyruvate kinase in the presence of trehalose. Arch Biochem Biophys. 2009;490(2):129–136.
- Henley JP, Sadana A. Deactivation theory. Biotechnol Bioeng. 1986;28(8):1277–1285.
- Jain NK, Roy I. Effect of trehalose on protein structure. Protein Sci. 2009;18(1):24–36.
- Ohtake S, Wang YJ. Trehalose: current use and future applications. J Pharm Sci. 2011;100(6):2020–2053.
- Brown DM, Upcroft JA, Upcroft P. A H2O-producing NADH oxidase from the protozoan parasite Giardia duodenalis. Eur J Biochem. 1996;241(1):155–161.
- Wang L, Chong H, Jiang R. Comparison of alkyl hydroperoxide reductase and two water-forming NADH oxidases from Bacillus cereus ATCC 14579. Appl Microbiol Biotechnol. 2012;96(5):1265–1273.
- Jiang R, Riebel BR, Bommarius AS. Comparison of alkyl hydroperoxide reductase (AhpR) and water-forming NADH oxidase from Lactococcus lactis ATCC 19435. Adv Synth Catal. 2005;347(7–8):1139–1146.
- Schmidt HL, Stöcklein W, Danzer J, et al. Isolation and properties of an H2O-forming NADH oxidase from Streptococcus faecalis. Eur J Biochem. 1986;156(1):149–155.
- Matulis D, Kranz JK, Salemme FR, et al. Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry. 2005;44(13):5258–5266.
- Lountos GT, Jiang R, Wellborn WB, et al. The crystal structure of NAD(P)H oxidase from Lactobacillus sanfranciscensis: insights into the conversion of O2 into two water molecules by the flavoenzyme. Biochemistry. 2006;45(32):9648–9659.
- Ödman P, Wellborn WB, Bommarius AS. An enzymatic process to L-ketoglutarate from L-glutamate: the coupled system L-glutamate dehydrogenase/NADH oxidase. Tetrahedron: Asymmetry. 2004;15(18):2933–2937.
- Hummel W, Riebel B. Isolation and biochemical characterization of a new NADH oxidase from Lactobacillus brevis. Biotechnol Lett. 2003;25(1):51–54.
- Park JT, Hirano J-I, Thangavel V, et al. NAD(P)H oxidase v from Lactobacillus plantarum (NoxV) displays enhanced operational stability even in absence of reducing agents. J Mol Catal B: Enzym. 2011;71(3–4):159–165.
- Nowak C, Beer B, Pick A, et al. A water-forming NADH oxidase from Lactobacillus pentosus suitable for the regeneration of synthetic biomimetic cofactors. Front Microbiol. 2015;6:957.
- Higuchi M, Shimada M, Yamamoto Y, et al. Identification of two distinct NADH oxidases corresponding to H2O2-forming oxidase and H2O-forming oxidase induced in Streptococcus mutans. J Gen Microbiol. 1993;139(10):2343–2351.
- Lin YZ, Liang SJ, Zhou JM, et al. Comparison of inactivation and conformational changes of D-glyceraldehyde-3-phosphate dehydrogenase during thermal denaturation. Biochim Biophys Acta. 1990;1038(2):247–252.
- Elbein AD, Pan YT, Pastuszak I, et al. New insights on trehalose: a multifunctional molecule. Glycobiology. 2003;13(4):17R–27R.
- Schiraldi C, Di Lernia I, De Rosa M. Trehalose production: exploiting novel approaches. Trends Biotechnol. 2002;20(10):420–425.
- Carninci P, Nishiyama Y, Westover A, et al. Thermostabilization and thermoactivation of thermolabile enzymes by trehalose and its application for the synthesis of full length cDNA. Proc Natl Acad Sci USA. 1998;95(2):520–524.
- Yang Q, Domesle KJ, Wang F, et al. Rapid detection of Salmonella in food and feed by coupling loop-mediated isothermal amplification with bioluminescent assay in real-time. BMC Microbiol. 2016;16(1):112–121.