
Abstract
White clover (Trifolium repens L.) is an important legume forage, which is widely distributed in cool-season regions. Therefore, cold stress is a major environmental factor limiting its growth and production, while little is known about the its cold tolerance at molecular level. In the present study, we performed time-course RNA-seq analysis under cold stress. RNA-seq results suggested that genes associated with “oxidoreductase activity” and “transcription regulator activity” are more likely related to the response of white clover to cold stress. To identify the specific gene modules and the hub genes of white clover in response to cold stress, we applied weighted gene co-expression network analyses (WGCNA) to transcriptome data. We also found that gene modules that focus on protein kinase activity, DNA-binding transcription factor activity and oxidoreductase activity, are more likely to be involved in the response of white clover to cold stress. Especially, we identified several AP2/ERF TF genes and CDPK genes as pivotal genes in white clover in response to cold stress, which would provide helpful insights into the molecular mechanisms underlying the response of white clover to cold stress.
Introduction
Crops are often exposed to various unfavorable environmental conditions, such as cold stress, water deficit and soil salinity [Citation1]. Among these stresses, cold stress is one of the major factors, which seriously limits crops growth, development and their distribution [Citation2]. Therefore, crops have been selected to resist cold stress, and these genes are classified into two major functional classes. The first class is mainly associated with antioxidants, transporters, osmo-protectants and various functional proteins; they play important roles in protecting plants from the damage of cold stress [Citation3]. The second class is made up of genes associated with regulatory functions in cellular metabolism, including signal transduction and transcriptional regulation processes. There are transcription factors (TFs), such as AP2/ERF, WRKY, MYB and NAC families, which are well identified and characterized with critical roles in response to cold stress from several plants [Citation1]. The protein kinases involved in various signal transduction pathways also have important roles in response to cold stress. Such enzymes are, for example, calcium-dependent protein kinases (CDPKs/CPKs), mitogen-activated protein kinase (MAPK) cascades and receptor-like kinases (RLKs), with well documented functions in model plants [Citation4].
White clover (Trifolium repens L.) is a cool-season perennial legume species, which is globally grown in wide climates [Citation5,Citation6]. However, in temperate climates, cold stress, especially in regions with hard winter, is an important constraint for its production. Consequently, cold tolerance has been one of most important traits in white clover, which has been well characterized in some morphological, physiological, and genetic breeding researches [Citation7–9]. Nevertheless, as a non-model plant, there are few reports about cold stress or cold tolerance published. With the limitation of allotetraploid genome and high genetic heterozygosity, the molecular mechanisms of white clover involved in the response to cold stress are still poorly understood [Citation10,Citation11].
Recently, with the development of high-throughput sequencing technologies, RNA-seq has become a cost-effective technology for measuring gene expressional profiles in plants, particularly those of non-model plants. When gene expressional data are well acquired and accumulated, genetic regulation networks (GRN) can be developed to explore molecular mechanisms of biological complex systems, including abiotic stress. Several genetic network analysis methods have been developed for reconstructing GRN, from gene expressional profiles, metabolism data and other molecular data. Among these GRN methods, weighted gene co-expression network analysis (WGCNA) is a popular and effective tool in reconstructing GRN based gene expression data. This method is widely employed to investigate microarray data and RNA-seq data in animal, plant and human genetics research. These results have confirmed its powerful and preponderant support in genomics view of biological systems [Citation12,Citation13].
In the present study, to explore the molecular mechanism of cold tolerance in white clover, we performed time-course RNA-seq analysis in conditions of cold stress, and established the global RNA expression profiles under cold stress. Meanwhile, key models focusing on protein kinase activity, DNA-binding transcription factor activity and oxidoreductase activity, were identified and characterized based on WGCNA of time-course expressional profiles. Based on WGCNA analysis results, WRKY, AP2/ERF TFs and CDPK genes are characterized as the hub genes, which have further suggested their potential critical roles in the response to cold stress in white clover.
Materials and methods
Plant materials and growth conditions
Seeds of white clover cv. Haifa were purchased from Barenbrug China Ltd. Com. (Beijing, China). The seeds were germinated on filter paper, and transferred to pots with a mixture of ∼400 mL perlite and sand at 3:1 in volume per pot as previously described [Citation14]. About 10–15 seedlings were planted in each pot. The seedlings were grown in a chamber, with 14 h in light at 18 °C and 10 h in dark at 24 °C each day, and each pot was irrigated with 50 mL of 1/2 Hoagland solution every two days. After four weeks of growth, the chamber was set at 4 °C, and whole seedings were collected at 0 mins (control), 30 mins, 1 h, 3 h, 6 h, 12 h, 24 h and 72 h (in total eight time points). For each time point, we collected three samples, with five seedlings per sample, and all samples were separately flash frozen in liquid nitrogen and then stored at −80 °C.
Construction and sequencing of RNA-seq library
Total RNA was extracted from each sample by using the RNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s instructions as our previous study described [Citation14]. The RNA samples were sent to BGI-Shenzhen Co. Ltd. (Shenzhen, China). The libraries were constructed according to the manufacturer’s instructions with mRNA library preparation kit (MGI, Shenzhen, China). RNA-seq was performed on the BGI-Seq platform with BGI-Seq500 model, and 150-bp paired-end reads were generated [Citation15].
Sequence assembly and annotation of RNA-seq data
Adapter sequences and low qualitative reads were first removed from the raw sequence data, and the cleaned reads of all the samples were submitted to NCBI SRA database (see PRJNA781064, https://dataview.ncbi.nlm.nih.gov/object/PRJNA781064?reviewer=nt3k7rsf9jm1ohclsg9l91g3kk). The clean reads were then assembled de novo into contigs using Trinity with default parameters [Citation16], and the contigs were further clustered into unigenes using CORSET with default parameters as described [Citation17]. These white clover unigenes were BLAST-searched against the combined database of protein sequences from Arabidopsis thaliana, rice (Oryza sativa), soybean (Glycine max), Medicago truncatula and red clover (Trifolium pratense) using BLASTX program for functional annotation (E-value cutoff: 1e-5) [Citation18]. Then, white clover unigenes were assigned with functional annotations based on their corresponding homologs in the combined database, including Gene Ontology (GO) and KOG (EuKaryotic Orthologous Groups) annotations [Citation19,Citation20]. In addition, these unigenes were also scanned using the iTAK pipeline to identify transcription factors as described previously [Citation21].
Expression analysis of genes involved in response to cold stress
All the RNA-seq reads were mapped to white clover unigenes using Salmon as previously described, and their expressional levels were quantified by Salmon quant [Citation22]. The matrix with the information of the number of the reads mapped to each gene were imported into R platform, and differentially expressed genes were detected using the DESeq2 package with the adjustment of p-value lower than 0.05 and the absolute value of fold change larger than 2, as described previously [Citation23]. DEGs were clustered and plotted using the ggplot2 package, while the GO annotations of these DEGs were retrieved based on previous homlogs research results, and GO enrichment analysis were performed using topGO package on R platform [Citation24].
Weighted gene co-expression network analysis
The R package WGCNA was used to identify the modules of highly correlated genes based on expression levels produced by Salmon quant as described above [Citation22,Citation25]. First, the white clover unigenes were filtered based on their expression and variance using “mad” function, and 15 K unigenes were selected with high expression variation among these time points under cold stress. Then, the soft threshold value 9 was chosen by using the “pickSoftThreshold” function. The adjacency matrix was generated, and the topological overlap matrix (TOM) was generated by using the TOM similarity algorithm. White clover unigenes were hierarchically clustered into modules, and module-traits (eight time points were treated as traits) associations were estimated, high correlation modules were identified and selected. The GO annotations of genes in high correlation modules were retrieved and functional enrichment analyses were performed using the topGO package. Meanwhile, gene co-expressional network visualization and analysis for selected modules were performed using the Cytoscape software, hub genes or key genes in co-expressional network were selected for further analysis [Citation26].
Results and discussion
Sequencing and de novo assembly of white clover transcriptome
To establish the transcriptional profiles in response to cold stress, we performed time-course RNA-seq of white clover with three experimental replicates for each time point. Next-generation sequencing was used to sequence these cDNA libraries, and around 657 million reads were generated (Supplemental Table S1). The clean reads were submitted to the National Center for Biotechnology Information (NCBI) SRA database (see PRJNA781064, as described above). To establish a white clover reference transcriptome database, we performed de novo assembly of all reads by using Trinity, and eliminated redundancy with CORSET (default parameters were used for both software tools). Finally, we obtained 72,685 transcripts (unigenes) from the de novo assembly with the mean length 1,302 bp and the N50 value 1,773 bp ().
Table 1. Summary of de novo assembled white clover transcriptome.
To annotate these unique transcripts, they were used as queries in a BLAST search of a database established by combining the protein sequences of the well-studied plant organisms: Arabidopsis thaliana, rice (Oryza sativa), soybean (Glycine max), Medicago truncatula and red clover (Trifolium pratense). The results showed that about 75.8% transcripts (55,078 ones) have homologs with significant hits (E-value cutoff: 1e-05) in the database (Supplemental Figure S1), higher than our previous reports in alfalfa (69.5%) [Citation14] and daylily (62.6%) [Citation15]. This is better than our previous RNA-seq assembly sequences, maybe owing to more reads and suitable assembly pipeline. As expected, longer transcripts had more annotated homolog hits than short ones (Supplemental Figure S2), implying that the identities of the longer transcripts have greater confidence, which is consistent with previous reports in other plants [Citation14]. In addition, all reads were mapped to unigenes with a mean mapping rate of 81.06%, while the mapping to red clover cDNA was 68.77%, suggesting that our unigenes have high confidence, and potential application in white clover genomics.
Figure 1. GO annotation results of white clover unique transcripts.
GO terms are listed at the X-axis, and the numbers of transcripts grouped in the GO terms are shown on Y-axis. Red bars represent Biological processes; Green bars represent Molecular functions; and blue bars represent cellular components.
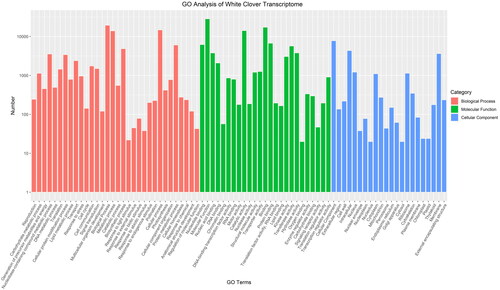
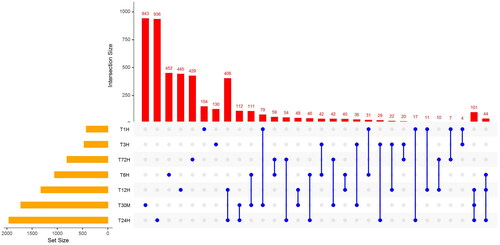
Figure 2. Differentially expressed genes in response to cold stress.
Time points and combinations of time-points are listed at the X-axis, and the numbers of differentially expressed genes are shown on Y-axis, while the orange bars are representing the numbers of DEG in each time points.
Functional annotation analysis of white clover transcripts
To determine the biological processes in which these 72,685 transcripts are involved, we further used GO terms and KOG annotations of their homologs in the well-studied model plant organisms to assign functions to these white clover transcripts. For GO term assignment, 32,642 genes (about 44.91%) were grouped into the following GO processes: transport (GO:0006810, 2398 unigenes), signal transduction (GO:0007165, 1503 unigenes), response to stress (GO:0007267, 964 unigenes), lipid metabolic process (GO:0006629, 793 unigenes), DNA metabolic process (GO:0006259, 494 unigenes) (). For KOG annotations, most KOG terms was classified into signal transduction mechanisms group (5,534 unigenes in 24,981 KOG annotation hits, 22.15%), general function prediction only (3,456 unigenes, 13.83%) and posttranslational modification, protein turnover and chaperones (2,529 unigenes, 10.12%), which are consistent with the GO annotations (Supplemental Figure S3). These annotation results provide valuable information for understanding the molecular mechanism of the response to cold stress in white clover. For example, the signal transduction process annotated from GO and KOG present most unigenes, implying their potential function under cold stress, and they were well and widely characterized with important roles in model plants with various stress, including cold stress [Citation4]. Meanwhile, transport, lipid metabolic processes are also considered as critical processes in response to cold stress, which have also been reported in previous research showing their response to cold stress [Citation2,Citation9]. Especially, unigenes involving the GO term ‘response to stress’ served as a direct clue for exploring the function of these unigenes in response to cold stress. In addition, 2906 plant transcript factor (TF) genes were identified and classified into 74 TF gene families according to the classification in PlantTFDB. The most abundant TF family was MYB (242 genes), followed by the AP2/ERF (236 genes), C2H2 (154 genes), WRKY (147 genes) and bHLH (133 genes) (Supplemental Figure S4). These TF families have been shown with key regulation functions in response to cold stress, which would be investigated in detail in future [Citation27].
Figure 3. Module-time-point associations with the application of WGCNA.
Each row corresponds to a module eigengene and each column to a time-point. Each cell contains the Pearson correlations and p-value, and the cells were colored based on Pearson correlation.
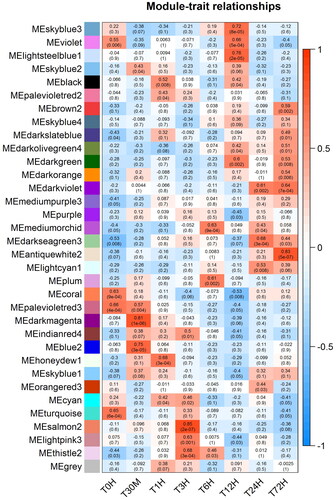
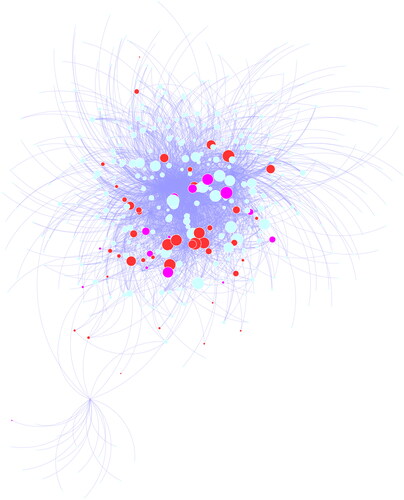
Figure 4. Gene networks in response to cold stress from the model MEdarkmagenta.
Modules of genes were exported from WGCNA results, and GRN were reconstructed in Cytoscape. Gene node size is proportional to number of edges (degree), red nodes correspond PK genes, pink nodes correspond TF genes, while blue nodes correspond other genes.
Differentially expressed genes in white clover response to cold stress
To identify differentially expressed genes (DEGs) in response to cold stress in white clover, we compared the gene expression levels between adjacent time points under cold stress using DESeq2 package on R platform, for example, 0 min vs. 30 min, 30 min vs. 1 h, etc. There were 5,273 genes differentially expressed (Fold change ≥2 or ≤ 0.5); T24H and T30M had most genes differentially expressed (). According to the GO annotations of these genes, they are highly enriched in “oxidoreductase activity” (GO:0016491, 421 genes, adjust p-value: 4.4e-27), implying that oxidoreductase related genes play critical roles in white clover in response to cold stress (Supplemental Table S2). These genes will activate reactive oxygen species (ROS), and increase antioxidant activity, which is well known as cold acclimation. Cold acclimation is a common process in many plants, and is used by a wide variety of plants to resist cold stress, which also works in white clover in response to cold stress [Citation3]. Other molecular functions such as, “transcription regulator activity” (GO:0140110, 156 genes, adjust p-value: 3.4e-20), and “metal ion binding” (GO:0046872, 328 genes, adjust p-value: 5.1e-09), were also significantly enriched with numerous members, and their functions have been considered as cold stress response related in other plants [Citation28]. For example, our data showed several transcript factors, including AP2/ERF, MYB, WRKY, NAC, etc, were characterized to function in response to cold stress. Similarly, metal ion binding was relative to phosphorylase kinase; for example, calmodulin is activated by the binding of Ca2+ ions, which is important to many cellar processes, including the response to cold stress [Citation29,Citation30]. Our results also confirmed these regulatory pathways or functions in white clover, implying their conservative function.
Table 2. GO enrichment analysis of genes from important modules.
WGCNA of white clover response to cold stress
To identify the specific gene modules and the hub genes of white clover in response to cold stress, we applied WGCNA to our RNA seq data. Based on parameter optimization, the soft-thresholding power of the adjacency matrix was set as nine. The adjacency matrix was calculated, and these genes were clustered and merged into 34 modules, and the correlations between gene modules and time points were calculated. For each time point, if the correlation with absolute value is larger than 0.5 (> 0.5 or < −0.5) and the p value is smaller than 0.01, it would be considered as significant module with effects in cold stress at corresponding time points (). Among these models, ten most significant modules were selected for further analysis (). These models are mainly related to DNA-binding transcription factor activity (GO:0003700), protein kinase activity (GO:0004672), oxidoreductase activity (GO:0016702) and autophagosome assembly (GO:0000045). These results were consisted with the function analysis of differentially expressed genes. Remarkably, the module MEdarkmagenta (cor: 0.81; p-value: 1e-06) is highly correlated with time point 30 M. This module includes 2208 genes that are mainly related to protein kinase activity (GO:0004672), DNA-binding transcription factor activity (GO:0003700) (). This finding suggests that genes involved in signal transduction process, including many TFs and PKs, played critical roles in response to cold stress, especially in the early responsive process. The genes in this module were selected to generate gene co-expression networks (GRN) with default parameters, and finally 473 genes and 2577 connections were obtained (). Ten hub genes with most gene links were all characterized as PK and TF, which also suggested their important function in response to cold stress. According to the gene annotation, most of them were characterized as RLK (32 members), CDPK (seven members), AP2/ERF (ten members) and WRKY (seven members), etc. When subjected to cold stress, white clover should perceive cold signals by cellar sensors, which were possibly RLKs and HKs emerging as key factors in cold signals transmission. In plant cells, they and protein kinases work together to initiate a transduction cascade; for example, cold stress will generate a Ca2+ signature by RLKs and HKs, which is named as a second messenger [Citation4]. The signal is decoded by numerous calmodulin relative proteins, such as calmodulin-like proteins, CDPKs and CBLs. The results showed seven CDPK genes present in early stage under cold stress, suggesting their pivotal function of the relevant signaling process in cold response. There were a number of function genes regulated by CDPK genes, which were well characterized in model plants. For example, two CRLK genes (Calcium/Calmodulin-Regulated Receptor-Like Kinases) perceive cold stress, and regulate the activity of MAPK cascades, which activates the expression of CBF, which are members of the AP2/ERF superfamily [Citation31]. The CBFs continue to regulation of downstream functional genes to enhance cold tolerance in Arabidopsis. It is worth noting that ten AP2/ERF TFs were also characterized in this gene model, which suggested that this regulation pathway is also present in white clover [Citation14]. RLKs and CDPKs receive the signals of cold stress, and transmit signals to key regulators, such as CBFs (AP2/ERF TFs), which activate downstream functional genes, including some TFs (WRKY, NAC, etc), and oxidoreductase, improving the cold tolerance of white clover.
Conclusions
In the present study, to investigate the molecular mechanism in white clover in response to cold stress, we performed time course RNA-seq analysis under cold stress. There were 5,273 genes identified as differentially expressed between different time points, which were mainly enriched in oxidoreductase activity and transcription regulator activity. The application of WGCNA to the transcriptome data revealed 34 modules with co-expression genes, including transcription factor, protein kinase and oxidoreductase, consistent with previous DEGs annotations. Meanwhile, genetic networks showed that CDPK, RLK, AP2/ERF and WRKY genes played important roles in the response to cold stress, which is helpful for understanding its molecular mechanism in white clover.
Author contributions
YS and CG conceived and the study, XZ, HY, and CC conducted the experiments, YS, XZ, HY, ML, YB and DG analyzed the data, YS and XZ wrote the manuscript, all authors read and approved the final manuscript.
Supplemental Material
Download PDF (833 KB)Data availability
The gene expression profiles and phenotype datasets of this study are provided as the Supporting Information Data Sheets S1, S2.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Knight MR, Knight H. Low-temperature perception leading to gene expression and cold tolerance in higher plants. New Phytol. 2012;195(4):737–751.
- Thomashow MF. Plant cold acclimation: freezing tolerance genes and regulatory mechanisms. Annu Rev Plant Physiol Plant Mol Biol. 1999;50(1):571–599.
- Qi J, Wang J, Gong Z, et al. Apoplastic ROS signaling in plant immunity. Curr Opin Plant Biol. 2017;38:92–100.
- Chen X, Ding Y, Yang Y, et al. Protein kinases in plant responses to drought, salt, and cold stress. J Integr Plant Biol. 2021;63(1):53–78.
- Collins RP, Helgadóttir Á, Frankow-Lindberg BE, et al. Temporal changes in population genetic diversity and structure in red and white clover grown in three contrasting environments in Northern Europe. Ann Bot. 2012;110(6):1341–1350.
- Wu F, Ma S, Zhou J, et al. Genetic diversity and population structure analysis in a large collection of white clover (Trifolium repens L.) germplasm worldwide. PeerJ. 2021;9:e11325. (
- Inostroza L, Bhakta M, Acuña H, et al. Understanding the complexity of cold tolerance in white clover using temperature gradient locations and a GWAS approach. Plant Genome. 2018;11(3):170096.
- Bao G, Tang W, He F, et al. Physiological response in the leaf and stolon of white clover under acid precipitation and freeze–thaw stress. Funct Plant Biol. 2019;47(1):50–57.
- Vaseva II, Anders I, Yuperlieva-Mateeva B, et al. Dehydrin expression as a potential diagnostic tool for cold stress in white clover. Plant Physiol Biochem. 2014;78:43–48.
- Griffiths AG, Moraga R, Tausen M, et al. Breaking free: the genomics of allopolyploidy-facilitated niche expansion in white clover. Plant Cell. 2019;31(7):1466–1487.
- Pu Q, Li Z, Nie G, et al. Selection and validation of reference genes for quantitative real-time PCR in white clover (Trifolium repens L.) involved in five abiotic stresses. Plants. 2020;9(8):996.
- Yu Y-H, Xin F, Dong L, et al. Weighted gene coexpression network analysis identifies critical genes in different subtypes of acute myeloid leukaemia. Biotechnol Biotechnol Equip. 2020;34(1):925–936.
- Li L-x, Qiao Z, Cai J-y, et al. Mineral element contents and gene expression in sophora tonkinensis during florescence. Biotechnol Biotechnol Equip. 2021;35(1):1593–1602.
- Shu Y, Li W, Zhao J, et al. Transcriptome sequencing analysis of alfalfa reveals CBF genes potentially playing important roles in response to freezing stress. Genet Mol Biol. 2017;40(4):824–833.
- Yang H, Zhang Y, Zhen X, et al. Transcriptome sequencing and expression profiling of genes involved in daylily (hemerocallis citrina borani) flower development. Biotechnol Biotechnol Equip. 2020;34(1):542–548.
- Haas BJ, Papanicolaou A, Yassour M, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8(8):1494–1512.
- Davidson NM, Oshlack A. Corset: enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014;15(7):410.
- Altschul SF, Madden TL, Schaffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402.
- Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;25(1):25–29.
- Tatusov RL, Fedorova ND, Jackson JD, et al. The COG database: an updated version includes eukaryotes. BMC Bioinf. 2003;4:41. (
- Jin J, Zhang H, Kong L, et al. PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2014;42(Database issue):D1182–7.
- Patro R, Duggal G, Love MI, et al. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14(4):417–419.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
- Alexa A, Rahnenfuhrer J. topGO: enrichment analysis for gene ontology. R package. 2019; version 2.38.1.(
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9(1):559.
- Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504.
- Gehan MA, Greenham K, Mockler TC, et al. Transcriptional networks-crops, clocks, and abiotic stress. Curr Opin Plant Biol. 2015;24:39–46. (
- Sharma R, Singh G, Bhattacharya S, et al. Comparative transcriptome Meta-analysis of Arabidopsis thaliana under drought and cold stress. PLoS One. 2018;13(9):e0203266.
- Yuan P, Yang T, Poovaiah BW. Calcium signaling-mediated plant response to cold stress. IJMS. 2018;19(12):3896.
- Atif RM, Shahid L, Waqas M, et al. Insights on calcium-dependent protein kinases (CPKs) signaling for abiotic stress tolerance in plants. IJMS. 2019;20(21):5298.
- Liu Z, Jia Y, Ding Y, et al. Plasma membrane CRPK1-Mediated phosphorylation of 14-3-3 proteins induces their nuclear import to Fine-Tune CBF signaling during cold response. Mol Cell. 2017;66(1):117–128.e5.